Sarcomeric Gene Variants and Their Role with Left Ventricular Dysfunction in Background of Coronary Artery Disease



Abstract
:1. Introduction:
2. Common Sarcomeric Protein and Associated Gene Polymorphism
3. Sarcomeric Proteins
3.1. Myofilament Proteins
3.1.1. Myosin
3.1.2. Actin
3.1.3. Myospryn
3.2. Regulatory Proteins
3.2.1. Tropomyosin (Tm)
3.2.2. Troponins
3.3. Sarcomeric Cytoskeletal Proteins
3.3.1. Titin Protein and Associated Gene Polymorphism
3.3.2. Myosin-Binding Protein C (Mybp-C) and Associated Gene Polymorphism
4. Common Sarcomeric Variants Reported with LVD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ghai, A.; Silversides, C.; Harris, L.; Webb, G.D.; Siu, S.C.; Therrien, J. Left ventricular dysfunction is a risk factor for sudden cardiac death in adults late after repair of tetralogy of Fallot. J. Am. Coll. Cardiol. 2002, 40, 1675–1680. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.; Ezekowitz, J.A.; Lewis, B.S.; Gersh, B.J.; van Diepen, S.; Amerena, J.; Bartunek, J.; Commerford, P.; Oh, B.H.; Harjola, V.P.; et al. Left ventricular systolic dysfunction, heart failure, and the risk of stroke and systemic embolism in patients with atrial fibrillation: Insights from the ARISTOTLE trial. Circ. Heart Fail. 2013, 6, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e272–e391. [Google Scholar]
- Stevens, S.M.; Reinier, K.; Chugh, S.S. Increased left ventricular mass as a predictor of sudden cardiac death: Is it time to put it to the test? Circ. Arrhythm. Electrophysiol. 2013, 6, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chonchol, M.; Goldenberg, I.; Moss, A.J.; McNitt, S.; Cheung, A.K. Risk factors for sudden cardiac death in patients with chronic renal insufficiency and left ventricular dysfunction. Am. J. Nephrol. 2007, 27, 7–14. [Google Scholar] [CrossRef]
- Benito, B.; Josephson, M.E. Ventricular tachycardia in coronary artery disease. Rev. Esp. Cardiol. 2012, 65, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Brun, F.; Spezzacatene, A.; Sinagra, G.; Taylor, M.R. Genetic Causes of Dilated Cardiomyopathy. Prog. Pediatr. Cardiol. 2014, 37, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Favalli, V.; Serio, A.; Grasso, M.; Arbustini, E. Genetic causes of dilated cardiomyopathy. Heart 2016, 102, 2004–2014. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Mattos, B.P.; Scolari, F.L.; Torres, M.A.; Simon, L.; Freitas, V.C.; Giugliani, R.; Matte, U. Prevalence and Phenotypic Expression of Mutations in the MYH7, MYBPC3 and TNNT2 Genes in Families with Hypertrophic Cardiomyopathy in the South of Brazil: A Cross-Sectional Study. Arq. Bras. Cardiol. 2016, 107, 257–265. [Google Scholar] [CrossRef]
- Rafael, J.F.; Cruz, F.F.; Carvalho, A.C.C.; Gottlieb, I.; Cazelli, J.G.; Siciliano, A.P.; Dias, G.M. Myosin-binding Protein C Compound Heterozygous Variant Effect on the Phenotypic Expression of Hypertrophic Cardiomyopathy. Arq. Bras. Cardiol. 2017, 108, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, A. Molecular basis of hereditary cardiomyopathy: Abnormalities in calcium sensitivity, stretch response, stress response and beyond. J. Hum. Genet. 2010, 55, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Dhandapany, P.S.; Sadayappan, S.; Xue, Y.; Powell, G.T.; Rani, D.S.; Nallari, P.; Rai, T.S.; Khullar, M.; Soares, P.; Bahl, A.; et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 2009, 41, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Mishra, A.; Srivastava, A.; Bhatt, M.; Garg, N.; Agarwal, S.K.; Pande, S.; Mittal, B. Role of common sarcomeric gene polymorphisms in genetic susceptibility to left ventricular dysfunction. J. Genet. 2016, 95, 263–272. [Google Scholar] [CrossRef]
- Srivastava, A.; Garg, N.; Mittal, T.; Khanna, R.; Gupta, S.; Seth, P.K.; Mittal, B. Association of 25 bp deletion in MYBPC3 gene with left ventricle dysfunction in coronary artery disease patients. PLoS ONE 2011, 6, e24123. [Google Scholar] [CrossRef]
- Tanjore, R.R.; Rangaraju, A.; Kerkar, P.G.; Calambur, N.; Nallari, P. MYBPC3 gene variations in hypertrophic cardiomyopathy patients in India. Can. J. Cardiol. 2008, 24, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Waldmuller, S.; Sakthivel, S.; Saadi, A.V.; Selignow, C.; Rakesh, P.G.; Golubenko, M.; Joseph, P.K.; Padmakumar, R.; Richard, P.; Schwartz, K.; et al. Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2003, 35, 623–636. [Google Scholar] [CrossRef]
- Rani, D.S.; Nallari, P.; Dhandapany, P.S.; Tamilarasi, S.; Shah, A.; Archana, V.; AshokKumar, M.; Narasimhan, C.; Singh, L.; Thangaraj, K. Cardiac Troponin T (TNNT2) mutations are less prevalent in Indian hypertrophic cardiomyopathy patients. DNA Cell Biol. 2012, 31, 616–624. [Google Scholar] [CrossRef]
- Komamura, K.; Iwai, N.; Kokame, K.; Yasumura, Y.; Kim, J.; Yamagishi, M.; Morisaki, T.; Kimura, A.; Tomoike, H.; Kitakaze, M.; et al. The role of a common TNNT2 polymorphism in cardiac hypertrophy. J. Hum. Genet. 2004, 49, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Bang, M.L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Granzier, H.L.; Radke, M.H.; Peng, J.; Westermann, D.; Nelson, O.L.; Rost, K.; King, N.M.; Yu, Q.; Tschope, C.; McNabb, M.; et al. Truncation of titin’s elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ. Res. 2009, 105, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagami, H.; Kikuchi, Y.; Katsuya, T.; Morishita, R.; Akasaka, H.; Saitoh, S.; Rakugi, H.; Kaneda, Y.; Shimamoto, K.; Ogihara, T. Gene polymorphism of myospryn (cardiomyopathy-associated 5) is associated with left ventricular wall thickness in patients with hypertension. Hypertens. Res. 2007, 30, 1239–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, C.C.; Trombitas, K.; Centner, T.; Kolmerer, B.; Stier, G.; Kunke, K.; Suzuki, K.; Obermayr, F.; Herrmann, B.; Granzier, H.; et al. The NH2 terminus of titin spans the Z-disc: Its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 1998, 143, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin diversity--alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef] [Green Version]
- Dos Remedios, C.; Gilmour, D. An historical perspective of the discovery of titin filaments. Biophys. Rev. 2017, 9, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Morales, N.; Holenka, T.K.; Schock, F. Filamin actin-binding and titin-binding fulfill distinct functions in Z-disc cohesion. PLoS Genet. 2017, 13, e1006880. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef]
- Tskhovrebova, L.; Trinick, J. Roles of titin in the structure and elasticity of the sarcomere. J. Biomed. Biotechnol. 2010, 2010, 612482. [Google Scholar] [CrossRef] [Green Version]
- Rall, J.A. What makes skeletal muscle striated? Discoveries in the endosarcomeric and exosarcomeric cytoskeleton. Adv. Physiol. Educ. 2018, 42, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Rayment, I.; Holden, H.M.; Whittaker, M.; Yohn, C.B.; Lorenz, M.; Holmes, K.C.; Milligan, R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science 1993, 261, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi-Takihara, K.; Sole, M.J.; Liew, J.; Ing, D.; Liew, C.C. Characterization of human cardiac myosin heavy chain genes. Proc. Natl. Acad. Sci. USA 1989, 86, 3504–3508. [Google Scholar] [CrossRef] [Green Version]
- Holubarsch, C.; Goulette, R.P.; Litten, R.Z.; Martin, B.J.; Mulieri, L.A.; Alpert, N.R. The economy of isometric force development, myosin isoenzyme pattern and myofibrillar ATPase activity in normal and hypothyroid rat myocardium. Circ. Res. 1985, 56, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.L.; Leinwand, L.A. Postnatal myosin heavy chain isoform expression in normal mice and mice null for IIb or IId myosin heavy chains. Dev. Biol. 2001, 229, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Lowes, B.D.; Minobe, W.; Abraham, W.T.; Rizeq, M.N.; Bohlmeyer, T.J.; Quaife, R.A.; Roden, R.L.; Dutcher, D.L.; Robertson, A.D.; Voelkel, N.F.; et al. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J. Clin. Invest. 1997, 100, 2315–2324. [Google Scholar] [CrossRef]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Lompre, A.M.; Schwartz, K.; d’Albis, A.; Lacombe, G.; Van Thiem, N.; Swynghedauw, B. Myosin isoenzyme redistribution in chronic heart overload. Nature 1979, 282, 105–107. [Google Scholar] [CrossRef]
- Swynghedauw, B. Developmental and functional adaptation of contractile proteins in cardiac and skeletal muscles. Physiol. Rev. 1986, 66, 710–771. [Google Scholar] [CrossRef]
- Takahashi, T.; Schunkert, H.; Isoyama, S.; Wei, J.Y.; Nadal-Ginard, B.; Grossman, W.; Izumo, S. Age-related differences in the expression of proto-oncogene and contractile protein genes in response to pressure overload in the rat myocardium. J. Clin. Invest. 1992, 89, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Herron, T.J.; McDonald, K.S. Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ. Res. 2002, 90, 1150–1152. [Google Scholar] [CrossRef] [Green Version]
- Reiser, P.J.; Portman, M.A.; Ning, X.H.; Schomisch Moravec, C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1814–H1820. [Google Scholar] [CrossRef] [Green Version]
- Eiras, S.; Narolska, N.A.; van Loon, R.B.; Boontje, N.M.; Zaremba, R.; Jimenez, C.R.; Visser, F.C.; Stooker, W.; van der Velden, J.; Stienen, G.J. Alterations in contractile protein composition and function in human atrial dilatation and atrial fibrillation. J. Mol. Cell Cardiol. 2006, 41, 467–477. [Google Scholar] [CrossRef]
- Meredith, C.; Herrmann, R.; Parry, C.; Liyanage, K.; Dye, D.E.; Durling, H.J.; Duff, R.M.; Beckman, K.; de Visser, M.; van der Graaff, M.M.; et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am. J. Hum. Genet. 2004, 75, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Darin, N.; Tajsharghi, H.; Ostman-Smith, I.; Gilljam, T.; Oldfors, A. New skeletal myopathy and cardiomyopathy associated with a missense mutation in MYH7. Neurology 2007, 68, 2041–2042. [Google Scholar] [CrossRef]
- Suurmeijer, A.J.; Clement, S.; Francesconi, A.; Bocchi, L.; Angelini, A.; Van Veldhuisen, D.J.; Spagnoli, L.G.; Gabbiani, G.; Orlandi, A. Alpha-actin isoform distribution in normal and failing human heart: A morphological, morphometric, and biochemical study. J. Pathol. 2003, 199, 387–397. [Google Scholar] [CrossRef]
- Kumar, A.; Crawford, K.; Flick, R.; Klevitsky, R.; Lorenz, J.N.; Bove, K.E.; Robbins, J.; Lessard, J.L. Transgenic overexpression of cardiac actin in the mouse heart suggests coregulation of cardiac, skeletal and vascular actin expression. Transgenic Res. 2004, 13, 531–540. [Google Scholar] [CrossRef]
- Boheler, K.R.; Carrier, L.; de la Bastie, D.; Allen, P.D.; Komajda, M.; Mercadier, J.J.; Schwartz, K. Skeletal actin mRNA increases in the human heart during ontogenic development and is the major isoform of control and failing adult hearts. J. Clin. Invest. 1991, 88, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.A.; Tinsley, C.L.; Blake, D.J. Myospryn is a novel binding partner for dysbindin in muscle. J. Biol. Chem. 2004, 279, 10450–10458. [Google Scholar] [CrossRef] [Green Version]
- Durham, J.T.; Brand, O.M.; Arnold, M.; Reynolds, J.G.; Muthukumar, L.; Weiler, H.; Richardson, J.A.; Naya, F.J. Myospryn is a direct transcriptional target for MEF2A that encodes a striated muscle, alpha-actinin-interacting, costamere-localized protein. J. Biol. Chem. 2006, 281, 6841–6849. [Google Scholar] [CrossRef] [Green Version]
- Kouloumenta, A.; Mavroidis, M.; Capetanaki, Y. Proper perinuclear localization of the TRIM-like protein myospryn requires its binding partner desmin. J. Biol. Chem. 2007, 282, 35211–35221. [Google Scholar] [CrossRef] [Green Version]
- Kielbasa, O.M.; Reynolds, J.G.; Wu, C.L.; Snyder, C.M.; Cho, M.Y.; Weiler, H.; Kandarian, S.; Naya, F.J. Myospryn is a calcineurin-interacting protein that negatively modulates slow-fiber-type transformation and skeletal muscle regeneration. FASEB J. 2011, 25, 2276–2286. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Kazzaz, J.A.; Helfman, D.M. Functional properties of non-muscle tropomyosin isoforms. Curr. Opin. Cell Biol. 1994, 6, 96–104. [Google Scholar] [CrossRef]
- Janco, M.; Suphamungmee, W.; Li, X.; Lehman, W.; Lehrer, S.S.; Geeves, M.A. Polymorphism in tropomyosin structure and function. J. Muscle Res. Cell Motil. 2013, 34, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Yumoto, F.; Ohki, S.Y.; Yasunaga, T.; Tanokura, M.; Wakabayashi, T. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J. Mol. Biol. 2005, 352, 178–201. [Google Scholar] [CrossRef]
- Pathan-Chhatbar, S.; Taft, M.H.; Reindl, T.; Hundt, N.; Latham, S.L.; Manstein, D.J. Three mammalian tropomyosin isoforms have different regulatory effects on nonmuscle myosin-2B and filamentous beta-actin in vitro. J. Biol. Chem. 2018, 293, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.V. Vertebrate tropomyosin: Distribution, properties and function. J. Muscle Res. Cell Motil. 2001, 22, 5–49. [Google Scholar] [CrossRef]
- Denz, C.R.; Narshi, A.; Zajdel, R.W.; Dube, D.K. Expression of a novel cardiac-specific tropomyosin isoform in humans. Biochem. Biophys. Res. Commun. 2004, 320, 1291–1297. [Google Scholar] [CrossRef]
- Purcell, I.F.; Bing, W.; Marston, S.B. Functional analysis of human cardiac troponin by the in vitro motility assay: Comparison of adult, foetal and failing hearts. Cardiovasc. Res. 1999, 43, 884–891. [Google Scholar] [CrossRef]
- Karam, C.N.; Warren, C.M.; Rajan, S.; de Tombe, P.P.; Wieczorek, D.F.; Solaro, R.J. Expression of tropomyosin-kappa induces dilated cardiomyopathy and depresses cardiac myofilament tension by mechanisms involving cross-bridge dependent activation and altered tropomyosin phosphorylation. J. Muscle Res. Cell Motil. 2011, 31, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Jagatheesan, G.; Karam, C.N.; Alves, M.L.; Bodi, I.; Schwartz, A.; Bulcao, C.F.; D’Souza, K.M.; Akhter, S.A.; Boivin, G.P.; et al. Molecular and functional characterization of a novel cardiac-specific human tropomyosin isoform. Circulation 2010, 121, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Filatov, V.L.; Katrukha, A.G.; Bulargina, T.V.; Gusev, N.B. Troponin: Structure, properties, and mechanism of functioning. Biochemistry 1999, 64, 969–985. [Google Scholar] [PubMed]
- Wilkinson, J.M.; Grand, R.J. Comparison of amino acid sequence of troponin I from different striated muscles. Nature 1978, 271, 31–35. [Google Scholar] [CrossRef]
- Wade, R.; Eddy, R.; Shows, T.B.; Kedes, L. cDNA sequence, tissue-specific expression, and chromosomal mapping of the human slow-twitch skeletal muscle isoform of troponin I. Genomics 1990, 7, 346–357. [Google Scholar] [CrossRef]
- Sasse, S.; Brand, N.J.; Kyprianou, P.; Dhoot, G.K.; Wade, R.; Arai, M.; Periasamy, M.; Yacoub, M.H.; Barton, P.J. Troponin I gene expression during human cardiac development and in end-stage heart failure. Circ. Res. 1993, 72, 932–938. [Google Scholar] [CrossRef] [Green Version]
- Fentzke, R.C.; Buck, S.H.; Patel, J.R.; Lin, H.; Wolska, B.M.; Stojanovic, M.O.; Martin, A.F.; Solaro, R.J.; Moss, R.L.; Leiden, J.M. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J. Physiol. 1999, 517((Pt. 1)), 143–157. [Google Scholar] [CrossRef]
- Huang, X.; Pi, Y.; Lee, K.J.; Henkel, A.S.; Gregg, R.G.; Powers, P.A.; Walker, J.W. Cardiac troponin I gene knockout: A mouse model of myocardial troponin I deficiency. Circ. Res. 1999, 84, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.J.; Jin, J.P. Gene regulation, alternative splicing, and posttranslational modification of troponin subunits in cardiac development and adaptation: A focused review. Front. Physiol. 2014, 5, 165. [Google Scholar] [CrossRef] [Green Version]
- Samson, F.; Mesnard, L.; Mihovilovic, M.; Potter, T.G.; Mercadier, J.J.; Roses, A.D.; Gilbert, J.R. A new human slow skeletal troponin T (TnTs) mRNA isoform derived from alternative splicing of a single gene. Biochem. Biophys. Res. Commun. 1994, 199, 841–847. [Google Scholar] [CrossRef]
- Anderson, P.A.; Malouf, N.N.; Oakeley, A.E.; Pagani, E.D.; Allen, P.D. Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ. Res. 1991, 69, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Craig, R.; Offer, G. The location of C-protein in rabbit skeletal muscle. Proc. R Soc. Lond. B. Biol. Sci. 1976, 192, 451–461. [Google Scholar] [PubMed]
- Wakabayashi, T. Mechanism of the calcium-regulation of muscle contraction--in pursuit of its structural basis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 321–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.D.; Ji, Y.T.; Zhou, X.H.; Li, H.L.; Zhang, H.T.; Xing, Q.; Hong, Y.F.; Tang, B.P. TNNT2 Gene Polymorphisms are Associated with Susceptibility to Idiopathic Dilated Cardiomyopathy in Kazak and Han Chinese. Med. Sci. Monit. 2015, 21, 3343–3347. [Google Scholar] [CrossRef] [Green Version]
- Ripoll-Vera, T.; Gamez, J.M.; Govea, N.; Gomez, Y.; Nunez, J.; Socias, L.; Escandell, A.; Rosell, J. Clinical and Prognostic Profiles of Cardiomyopathies Caused by Mutations in the Troponin T Gene. Rev. Esp. Cardiol. 2016, 69, 149–158. [Google Scholar] [CrossRef]
- Hein, S.; Kostin, S.; Heling, A.; Maeno, Y.; Schaper, J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000, 45, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Whiting, A.; Wardale, J.; Trinick, J. Does titin regulate the length of muscle thick filaments? J. Mol. Biol. 1989, 205, 263–268. [Google Scholar] [CrossRef]
- Miller, M.K.; Granzier, H.; Ehler, E.; Gregorio, C.C. The sensitive giant: The role of titin-based stretch sensing complexes in the heart. Trends Cell Biol. 2004, 14, 119–126. [Google Scholar] [CrossRef]
- LeWinter, M.M.; Granzier, H.L. Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 2014, 63, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Itoh-Satoh, M.; Hayashi, T.; Nishi, H.; Koga, Y.; Arimura, T.; Koyanagi, T.; Takahashi, M.; Hohda, S.; Ueda, K.; Nouchi, T.; et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 291, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Siu, B.L.; Niimura, H.; Osborne, J.A.; Fatkin, D.; MacRae, C.; Solomon, S.; Benson, D.W.; Seidman, J.G.; Seidman, C.E. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation 1999, 99, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Pleitner, J.M.; Saupe, K.W.; Greaser, M.L. Pathophysiological defects and transcriptional profiling in the RBM20-/- rat model. PLoS ONE 2013, 8, e84281. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [Green Version]
- Flashman, E.; Redwood, C.; Moolman-Smook, J.; Watkins, H. Cardiac myosin binding protein C: Its role in physiology and disease. Circ. Res. 2004, 94, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Kensler, R.W.; Craig, R.; Moss, R.L. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc. Natl. Acad. Sci. USA 2017, 114, E1355–E1364. [Google Scholar] [CrossRef] [Green Version]
- Mamidi, R.; Gresham, K.S.; Verma, S.; Stelzer, J.E. Cardiac Myosin Binding Protein-C Phosphorylation Modulates Myofilament Length-Dependent Activation. Front. Physiol. 2016, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Weber, F.E.; Vaughan, K.T.; Reinach, F.C.; Fischman, D.A. Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment. Eur. J. Biochem. 1993, 216, 661–669. [Google Scholar] [CrossRef]
- Gautel, M.; Zuffardi, O.; Freiburg, A.; Labeit, S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: A modulator of cardiac contraction? EMBO J. 1995, 14, 1952–1960. [Google Scholar] [CrossRef]
- Dhoot, G.K.; Hales, M.C.; Grail, B.M.; Perry, S.V. The isoforms of C protein and their distribution in mammalian skeletal muscle. J. Muscle Res. Cell Motil. 1985, 6, 487–505. [Google Scholar] [CrossRef]
- Reinach, F.C.; Masaki, T.; Fischman, D.A. Characterization of the C-protein from posterior latissimus dorsi muscle of the adult chicken: Heterogeneity within a single sarcomere. J. Cell Biol. 1983, 96, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M.; Furst, D.O.; Cocco, A.; Schiaffino, S. Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: Lack of isoform transcomplementation in cardiac muscle. Circ. Res. 1998, 82, 124–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.; Robbins, J. Signaling and myosin-binding protein C. J. Biol. Chem 2011, 286, 9913–9919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadayappan, S.; de Tombe, P.P. Cardiac myosin binding protein-C as a central target of cardiac sarcomere signaling: A special mini review series. Pflugers Arch. 2014, 466, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Sarikas, A.; Carrier, L.; Schenke, C.; Doll, D.; Flavigny, J.; Lindenberg, K.S.; Eschenhagen, T.; Zolk, O. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc. Res. 2005, 66, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.L.; Szweda, L.I.; Friguet, B. Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 2002, 397, 298–304. [Google Scholar] [CrossRef]
- Okada, K.; Wangpoengtrakul, C.; Osawa, T.; Toyokuni, S.; Tanaka, K.; Uchida, K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 1999, 274, 23787–23793. [Google Scholar] [CrossRef] [Green Version]
- Simonson, T.S.; Zhang, Y.; Huff, C.D.; Xing, J.; Watkins, W.S.; Witherspoon, D.J.; Woodward, S.R.; Jorde, L.B. Limited distribution of a cardiomyopathy-associated variant in India. Ann. Hum. Genet. 2010, 74, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Anand, A.; Chin, C.; Shah, A.S.V.; Kwiecinski, J.; Vesey, A.; Cowell, J.; Weber, E.; Kaier, T.; Newby, D.E.; Dweck, M.; et al. Cardiac myosin-binding protein C is a novel marker of myocardial injury and fibrosis in aortic stenosis. Heart 2018, 104, 1101–1108. [Google Scholar] [CrossRef]
- Mishra, A.; Srivastava, A.; Mittal, T.; Garg, N.; Mittal, B. Impact of renin-angiotensin-aldosterone system gene polymorphisms on left ventricular dysfunction in coronary artery disease patients. Dis. Markers 2012, 32, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Farza, H.; Townsend, P.J.; Carrier, L.; Barton, P.J.; Mesnard, L.; Bahrend, E.; Forissier, J.F.; Fiszman, M.; Yacoub, M.H.; Schwartz, K. Genomic organisation, alternative splicing and polymorphisms of the human cardiac troponin T gene. J. Mol. Cell Cardiol. 1998, 30, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mishra, A.; Srivastava, A.; Mittal, T.; Garg, N.; Mittal, B. Significant role of ADRB3 rs4994 towards the development of coronary artery disease. Coron. Artery Dis. 2014, 25, 29–34. [Google Scholar] [CrossRef]
- Mishra, A.; Srivastava, A.; Mittal, T.; Garg, N.; Mittal, B. Genetic predisposition to left ventricular dysfunction: A multigenic and multi-analytical approach. Gene 2014, 546, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Sadayappan, S.; Osinska, H.; Klevitsky, R.; Lorenz, J.N.; Sargent, M.; Molkentin, J.D.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA 2006, 103, 16918–16923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Srivastava, A.; Mittal, T.; Garg, N.; Mittal, B. Association of matrix metalloproteinases (MMP2, MMP7 and MMP9) genetic variants with left ventricular dysfunction in coronary artery disease patients. Clin. Chim. Acta 2012, 413, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Srivastava, A.; Mittal, T.; Garg, N.; Mittal, B. Role of inflammatory gene polymorphisms in left ventricular dysfunction (LVD) susceptibility in coronary artery disease (CAD) patients. Cytokine 2013, 61, 856–861. [Google Scholar] [CrossRef]
- Akasheva, D.U.; Plokhova, E.V.; Tkacheva, O.N.; Strazhesko, I.D.; Dudinskaya, E.N.; Kruglikova, A.S.; Pykhtina, V.S.; Brailova, N.V.; Pokshubina, I.A.; Sharashkina, N.V.; et al. Age-Related Left Ventricular Changes and Their Association with Leukocyte Telomere Length in Healthy People. PLoS ONE 2015, 10, e0135883. [Google Scholar] [CrossRef]
- Hayward, C.S.; Kalnins, W.V.; Kelly, R.P. Gender-related differences in left ventricular chamber function. Cardiovasc. Res. 2001, 49, 340–350. [Google Scholar] [CrossRef]
- Kishi, S.; Reis, J.P.; Venkatesh, B.A.; Gidding, S.S.; Armstrong, A.C.; Jacobs, D.R., Jr.; Sidney, S.; Wu, C.O.; Cook, N.L.; Lewis, C.E.; et al. Race-ethnic and sex differences in left ventricular structure and function: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. J. Am. Heart Assoc. 2015, 4, e001264. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, A.M.; Azoz, A.M.; Shaheen, H.A.; Farrag, Y.A.; Khalifa, M.A.; Youssef, A. Acute effects of cigarette smoking on the cardiac diastolic functions. J. Saudi Heart Assoc. 2013, 25, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triposkiadis, F.; Giamouzis, G.; Parissis, J.; Starling, R.C.; Boudoulas, H.; Skoularigis, J.; Butler, J.; Filippatos, G. Reframing the association and significance of co-morbidities in heart failure. Eur J. Heart Fail. 2016, 18, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Srivastava, A.; Kumar, S.; Mittal, T.; Garg, N.; Agarwal, S.K.; Pande, S.; Mittal, B. Role of angiotensin II type I (AT1 A1166C) receptor polymorphism in susceptibility of left ventricular dysfunction. Indian Heart J. 2015, 67, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
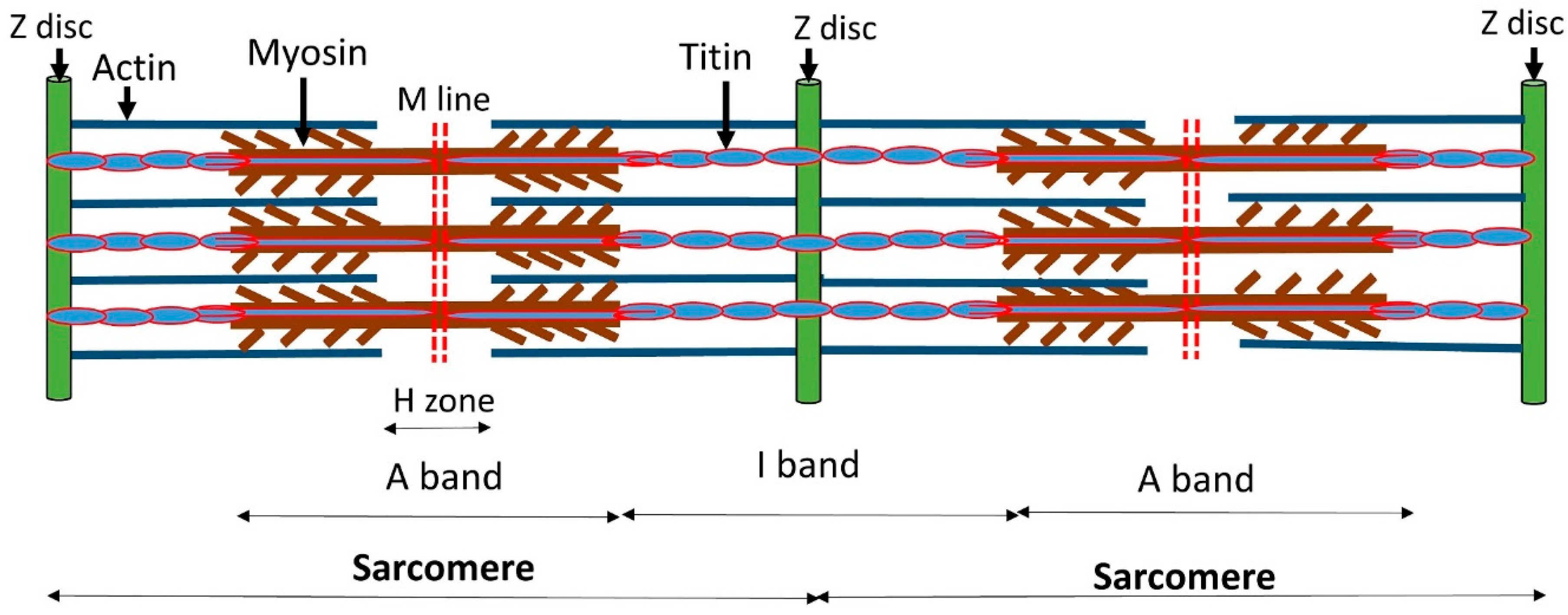
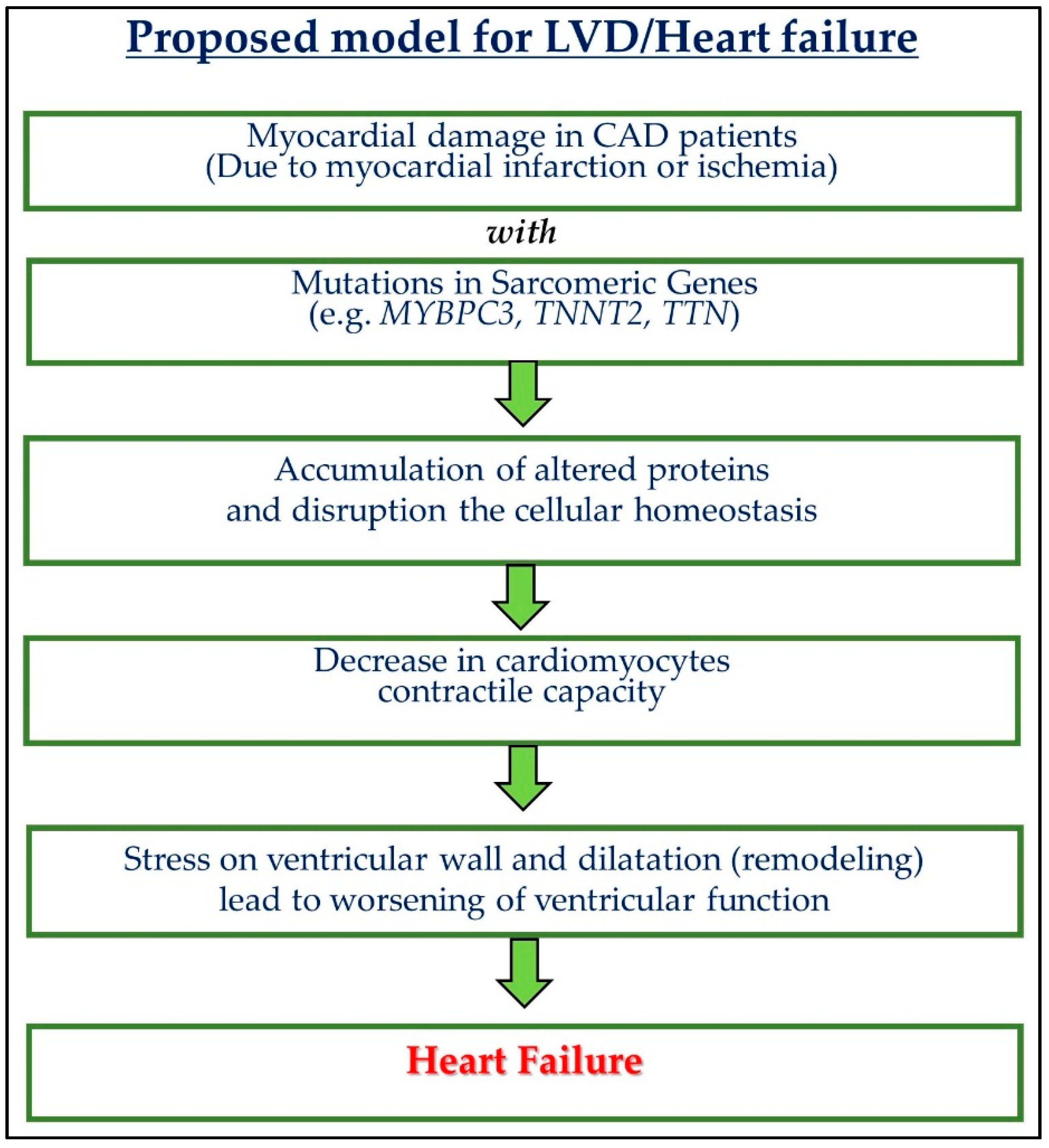

{kind=link}
{kind=link}
| Genes | Location | Type of Polymorphism | Functional Role | Ref. |
|---|---|---|---|---|
| MYBPC3 | 11p11.2 | 25 bp Ins/del | MYBPC3 gene mutation is associated with inherited cardiomyopathies and an increased heart failure risk | [14,15,16,17,18] |
| TNNT2 | 1q32 | 5 bp Ins/del | The 5 bp (CTTCT) deletion in intron 3 of the TNNT2 gene at the polypyrimidine tract was found to affect the gene splicing and branch site selection | [10,19,20] |
| TTN | 2q31 | 18 bp Ins/del | This deletion is present within the PEVK region of titin gene that regulates the extensibility of the protein | [21,22] |
| Myospryn | 5q14.1 | K2906N | This polymorphism is associated with cardiac adaptation in response to pressure overload, left ventricular hypertrophy, and left ventricular diastolic dysfunction in hypertensive patients | [23] |
| Common Factors | Effect | Ref. |
|---|---|---|
| Environmental Risk Factors | ||
| Age | Higher in older patients | [108] |
| Gender | More in men | [109] |
| Ethnicity | High in African Athletes | [110] |
| Smoking status | Higher in smoker patients | [111] |
| Obesity | Higher in obese patients | [112] |
| Hypertension | Higher in hypertensive patients | [112] |
| Coronary artery disease | Higher in CAD patients | [112] |
| Renal disease | Higher in CKD patients | [112] |
| Genetic Risk Factors | ||
| Sarcomeric gene mutations–MYBPC3, TNNT2, TTN, MYH7, Myospryn, etc. | ↑ ventricular remodeling and LVD | [15,23] |
| Renin–Angiotensin–Aldosterone System (RAAS) pathway–ACE and AT1 Gene | ↑ ventricular remodeling and LVD | [101,113] |
| Matrix Metalloproteinase (MMPs)–MMP2, MMP7 and MMP9 | ↑ LVD | [104,106] |
| Adrenergic pathway–ADRB1, ADRA2A, ADRB3 | ↑ ventricular remodeling and LVD | [103] |
| Inflammatory pathway–NFKB1, IL6, and TNF-α | ↑ ventricular remodeling and LVD | [104,107] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Kumar, V.; Kim, J.-J. Sarcomeric Gene Variants and Their Role with Left Ventricular Dysfunction in Background of Coronary Artery Disease. Biomolecules 2020, 10, 442. https://doi.org/10.3390/biom10030442
Kumar S, Kumar V, Kim J-J. Sarcomeric Gene Variants and Their Role with Left Ventricular Dysfunction in Background of Coronary Artery Disease. Biomolecules. 2020; 10(3):442. https://doi.org/10.3390/biom10030442
Chicago/Turabian StyleKumar, Surendra, Vijay Kumar, and Jong-Joo Kim. 2020. "Sarcomeric Gene Variants and Their Role with Left Ventricular Dysfunction in Background of Coronary Artery Disease" Biomolecules 10, no. 3: 442. https://doi.org/10.3390/biom10030442
APA StyleKumar, S., Kumar, V., & Kim, J.-J. (2020). Sarcomeric Gene Variants and Their Role with Left Ventricular Dysfunction in Background of Coronary Artery Disease. Biomolecules, 10(3), 442. https://doi.org/10.3390/biom10030442