Actionable Potentials of Less Frequently Mutated Genes in Colorectal Cancer and Their Roles in Precision Medicine

 , ,
, ,
Abstract
:1. Introduction
2. Less Frequently Mutated Genes in CRC
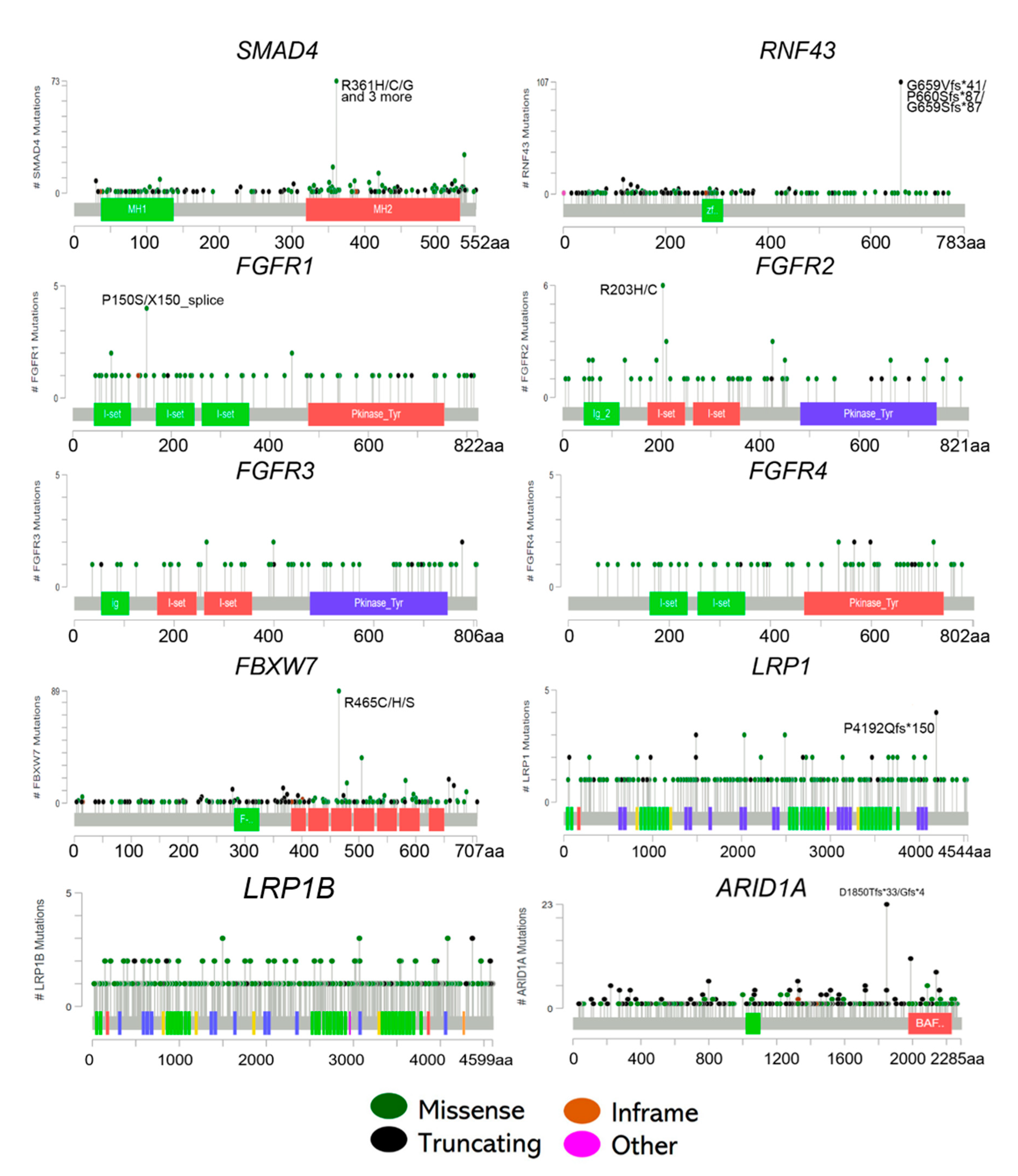
3. SMAD4 Mutations
4. RNF43 Mutations
5. FGFR Mutations
6. FBXW7 Mutations
7. LRP1 Mutations
8. ARID1A Mutations
9. Co-occurrence of the Less Frequently Mutated Genes
10. Other Genomic Alterations: Large Genomic Rearrangement and Deletions
11. Future Recommendations and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. Ca Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourhoseingholi, M.A. Increased burden of colorectal cancer in Asia. World J. Gastrointest. Oncol. 2012, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Pourhoseingholi, M.A. Epidemiology and burden of colorectal cancer in Asia-Pacific region: What shall we do now? Transl. Gastrointest. Cancer 2014, 3, 169–173. [Google Scholar]
- Van Der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.L.; Ricci, V.; Vivenza, D.; Granetto, C.; Fabozzi, T.; Miraglio, E.; Merlano, M.C. Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J. Gastroenterol. 2016, 22, 6944–6954. [Google Scholar] [CrossRef] [PubMed]
- Kamatham, S.; Shahjehan, F.; Kasi, P.M. Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer: Current Status, Recent Advances, and Future Directions. Curr. Colorectal. Cancer Rep. 2019, 15, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [Green Version]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Wu, G.; Wilson, G.; George, J.; Liddle, C.; Hebbard, L.; Qiao, L. Overcoming treatment resistance in cancer: Current understanding and tactics. Cancer Lett. 2017, 387, 69–76. [Google Scholar] [CrossRef]
- Sandhu, J.; Lavingia, V.; Fakih, M. Systemic treatment for metastatic colorectal cancer in the era of precision medicine. J. Surg. Oncol. 2019, 119, 564–582. [Google Scholar] [CrossRef] [PubMed]
- Rachiglio, A.M.; Lambiase, M.; Fenizia, F.; Roma, C.; Cardone, C.; Iannaccone, A.; De Luca, A.; Carotenuto, M.; Frezzetti, D.; Martinelli, E.; et al. Genomic Profiling of KRAS/NRAS/BRAF/PIK3CA Wild-Type Metastatic Colorectal Cancer Patients Reveals Novel Mutations in Genes Potentially Associated with Resistance to Anti-EGFR Agents. Cancers 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremolini, C.; Benelli, M.; Fontana, E.; Pagani, F.; Rossini, D.; Fucà, G.; Busico, A.; Conca, E.; Di Donato, S.; Loupakis, F.; et al. Benefit from anti-EGFRs in RAS and BRAF wild-type metastatic transverse colon cancer: A clinical and molecular proof of concept study. ESMO Open 2019, 4, e000489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Albéniz, X.; Alonso, V.; Escudero, P.; Méndez, M.; Gallego, J.; Rodríguez, J.R.; Salud, A.; Fernández-Plana, J.; Manzano, H.; Zanui, M.; et al. Prospective Biomarker Study in Advanced RAS Wild-Type Colorectal Cancer: POSIBA Trial (GEMCAD 10-02). Oncologist 2019, 24, e1115–e1122. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Maria, A.; Na, N.; da Cruz Paula, A.; Gorelick, A.N.; Hechtman, J.F.; Carson, J.; Lefkowitz, R.A.; Weigelt, B.; Taylor, B.S.; et al. V211D Mutation in MEK1 Causes Resistance to MEK Inhibitors in Colon Cancer. Cancer Discov. 2019, 9, 1182–1191. [Google Scholar] [CrossRef]
- Gbenedio, O.M.; Bonnans, C.; Grun, D.; Wang, C.-Y.; Hatch, A.J.; Mahoney, M.R.; Barras, D.; Matli, M.; Miao, Y.; Garcia, K.C.; et al. RasGRP1 is a potential biomarker to stratify anti-EGFR therapy response in colorectal cancer. Jci. Insight 2019, 5, 127552. [Google Scholar]
- Mao, C.; Wu, X.-Y.; Yang, Z.-Y.; Threapleton, D.E.; Yuan, J.-Q.; Yu, Y.-Y.; Tang, J.-L. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci. Rep. 2015, 5, 8065. [Google Scholar] [CrossRef]
- Dienstmann, R.; Tabernero, J. Spectrum of Gene Mutations in Colorectal Cancer Implications for Treatment. Cancer J. 2016, 22, 149–155. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Malapelle, U.; Pisapia, P.; Sgariglia, R.; Vigliar, E.; Biglietto, M.; Carlomagno, C.; Giuffrè, G.; Bellevicine, C.; Troncone, G. Less frequently mutated genes in colorectal cancer: Evidences from next-generation sequencing of 653 routine cases. J. Clin. Pathol. 2016, 69, 767–771. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [Green Version]
- Yu, J.; Wu, W.K.K.; Li, X.; He, J.; Li, X.-X.; Ng, S.S.M.; Yu, C.; Gao, Z.; Yang, J.; Li, M.; et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut 2015, 64, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Shao, Y.W.; Lin, P.; Cai, X.; Wang, B.; Ding, Y.; Ma, X.; Wu, X.; Xia, Y.; Zhu, D.; et al. SMAD4 and NF1 mutations as potential biomarkers for poor prognosis to cetuximab-based therapy in Chinese metastatic colorectal cancer patients. BMC Cancer 2018, 18, 479. [Google Scholar] [CrossRef] [Green Version]
- Sarshekeh, A.M.; Advani, S.; Overman, M.J.; Manyam, G.; Kee, B.K.; Fogelman, D.R.; Dasari, A.; Raghav, K.; Vilar, E.; Manuel, S.; et al. Association of SMAD4 mutation with patient demographics, tumor characteristics, and clinical outcomes in colorectal cancer. PLoS ONE 2017, 12, e0173345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Leng, C.; Wu, C.; Zhang, Z.; Dou, L.; Luo, X.; Zhang, B.; Chen, X.; Dou, L.; Dou, L.; et al. Smad4 sensitizes colorectal cancer to 5-fluorouracil through cell cycle arrest by inhibiting the PI3K/Akt/CDC2/survivin cascade. Oncol. Rep. 2016, 35, 1807–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Eto, T.; Miyake, K.; Nosho, K.; Ohmuraya, M.; Imamura, Y.; Arima, K.; Kanno, S.; Fu, L.; Kiyozumi, Y.; Izumi, D.; et al. Impact of loss-of-function mutations at the RNF43 locus on colorectal cancer development and progression. J. Pathol. 2018, 245, 445–455. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [Green Version]
- Tai, D.; Wells, K.; Arcaroli, J.; Vanderbilt, C.; Aisner, D.L.; Messersmith, W.A.; Lieu, C.H. Targeting the WNT Signaling Pathway in Cancer Therapeutics. Oncologist 2015, 20, 1189–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumeyer, V.; Grandl, M.; Dietl, A.; Brutau-Abia, A.; Allgäuer, M.; Kalali, B.; Zhang, Y.; Pan, K.-F.; Steiger, K.; Vieth, M.; et al. Loss of endogenous RNF43 function enhances proliferation and tumour growth of intestinal and gastric cells. Carcinogenesis 2019, 40, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.H.; Cottrell, C.E.; McNulty, S.N.; Vigh-Conrad, K.A.; Lamp, S.; Heusel, J.W.; Duncavage, E.J. FGFR2 amplification in colorectal adenocarcinoma. Cold Spring Harb Mol. Case Stud. 2017, 3, a001495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR landscape in cancer: Analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M.; et al. FGFR2 Gene Amplification in Gastric Cancer Predicts Sensitivity to the Selective FGFR Inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef] [Green Version]
- Mathur, A.; Ware, C.; Davis, L.; Gazdar, A.; Pan, B.-S.; Lutterbach, B. FGFR2 is amplified in the NCI-H716 colorectal cancer cell line and is required for growth and survival. PLoS ONE 2014, 9, e98515. [Google Scholar] [CrossRef]
- Mohd Yunos, R.-I.; Ab Mutalib, N.-S.; Sean, K.S.; Saidin, S.; Abdul Razak, M.R.; Mahamad Nadzir, N.; Abd Razak, Z.; Mohamed Rose, I.; Sagap, I.; Mazlan, L.; et al. Whole exome sequencing identifies genomic alterations in proximal and distal colorectal cancer. Prog. Microbes Mol. Biol. 2019, 2, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jardim, D.L.; Wheler, J.J.; Hess, K.; Tsimberidou, A.M.; Zinner, R.; Janku, F.; Subbiah, V.; Naing, A.; Piha-Paul, S.A.; Westin, S.N.; et al. FBXW7 mutations in patients with advanced cancers: Clinical and molecular characteristics and outcomes with mTOR inhibitors. PLoS ONE 2014, 9, e89388. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, H.H.; Lin, J.K.; Lin, C.C.; Lan, Y.T.; Wang, H.S.; Yang, S.H.; Chen, W.S.; Lin, T.C.; Jiang, J.K.; et al. FBXW7 mutation analysis and its correlation with clinicopathological features and prognosis in colorectal cancer patients. Int. J. Biol. Markers 2015, 30, e88–e95. [Google Scholar] [CrossRef]
- Abdul, S.-N.; Ab Mutalib, N.-S.; Sean, K.S.; Syafruddin, S.E.; Ishak, M.; Sagap, I.; Mazlan, L.; Rose, I.M.; Abu, N.; Mokhtar, N.M.; et al. Molecular Characterization of Somatic Alterations in Dukes’ B and C Colorectal Cancers by Targeted Sequencing. Front. Pharm. 2017, 8, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.-H.; Kim, I.-J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valliyammai, N.; Nancy, N.K.; Sagar, T.G.; Rajkumar, T. Study of NOTCH1 and FBXW7 Mutations and Its Prognostic Significance in South Indian T-Cell Acute Lymphoblastic Leukemia. J. Pediatr Hematol. Oncol. 2018, 40, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Korphaisarn, K.; Morris, V.K.; Overman, M.J.; Fogelman, D.R.; Kee, B.K.; Raghav, K.P.S.; Manuel, S.; Shureiqi, I.; Wolff, R.A.; Eng, C.; et al. FBXW7 missense mutation: A novel negative prognostic factor in metastatic colorectal adenocarcinoma. Oncotarget 2017, 8, 39268–39279. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Tan, S.; Zou, F.; Yu, J.; Zhang, L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017, 36, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Boulagnon-Rombi, C.; Schneider, C.; Leandri, C.; Jeanne, A.; Grybek, V.; Bressenot, A.M.; Barbe, C.; Marquet, B.; Nasri, S.; Coquelet, C.; et al. LRP1 expression in colon cancer predicts clinical outcome. Oncotarget 2018, 9, 8849–8869. [Google Scholar] [CrossRef] [Green Version]
- Salama, Y.; Lin, S.-Y.; Dhahri, D.; Hattori, K.; Heissig, B. The fibrinolytic factor tPA drives LRP1-mediated melanoma growth and metastasis. FASEB J. 2019, 33, 3465–3480. [Google Scholar] [CrossRef]
- Mehrvarz Sarshekeh, A.; Loree, J.M.; Manyam, G.C.; Pereira, A.A.L.; Raghav, K.P.S.; Lam, M.; Davis, J.S.; Dasari, A.; Morris, V.K.; Menter, D.; et al. The characteristics of ARID1A mutations in colorectal cancer. J. Clin. Oncol. 2018, 36, 3595. [Google Scholar] [CrossRef]
- Wei, X.-L.; Wang, D.-S.; Xi, S.-Y.; Wu, W.-J.; Chen, D.-L.; Zeng, Z.-L.; Wang, R.-Y.; Huang, Y.-X.; Jin, Y.; Wang, F.; et al. Clinicopathologic and prognostic relevance of ARID1A protein loss in colorectal cancer. World J. Gastroenterol. 2014, 20, 18404–18412. [Google Scholar] [CrossRef]
- Xie, C.; Fu, L.; Han, Y.; Li, Q.; Wang, E. Decreased ARID1A expression facilitates cell proliferation and inhibits 5-fluorouracil-induced apoptosis in colorectal carcinoma. Tumor Biol. 2014, 35, 7921–7927. [Google Scholar] [CrossRef]
- Niedermaier, B.; Sak, A.; Zernickel, E.; Xu, S.; Groneberg, M.; Stuschke, M. Targeting ARID1A-mutant colorectal cancer: Depletion of ARID1B increases radiosensitivity and modulates DNA damage response. Sci. Rep. 2019, 9, 18207. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming Growth Factor β Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFΒ 2 signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Petit, F.G.; Deng, C.; Jamin, S.P. Partial müllerian duct retention in Smad4 conditional mutant male mice. Int. J. Biol. Sci. 2016, 12, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Freeman, T.J.; Smith, J.J.; Chen, X.; Washington, M.K.; Roland, J.T.; Means, A.L.; Eschrich, S.A.; Yeatman, T.J.; Deane, N.G.; Beauchamp, R.D. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology 2012, 142, 562–571.e2. [Google Scholar] [CrossRef] [Green Version]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaki, M.; Iijima, T.; Konishi, M.; Sakai, K.; Ishii, A.; Yasuno, M.; Hishima, T.; Koike, M.; Shitara, N.; Iwama, T.; et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 1999, 18, 3098–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losi, L.; Bouzourene, H.; Benhattar, J. Loss of Smad4 expression predicts liver metastasis in human colorectal cancer. Oncol. Rep. 2007, 17, 1095–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaksson-Mettävainio, M.; Palmqvist, R.; Dahlin, A.M.; Van Guelpen, B.; Rutegård, J.; Öberg, Å.; Henriksson, M.L. High SMAD4 levels appear in microsatellite instability and hypermethylated colon cancers, and indicate a better prognosis. Int. J. Cancer 2012, 131, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-Y.; Lee, J.-A.; Shin, Y.; Cho, N.-Y.; Bae, J.M.; Kang, G.H. Clinicopathological Characterization and Prognostic Implication of SMAD4 Expression in Colorectal Carcinoma. J. Pathol. Transl. Med. 2019, 53, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Mokarram, P.; Azimi, H.; Olumi, H.; Varma, S.; Nickerson, M.L.; Brim, H. Targeted exome sequencing reveals distinct pathogenic variants in Iranians with colorectal cancer. Oncotarget 2016, 8, 7852–7866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulay, J.-L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy in patients with colorectal cancer. Br. J. Cancer 2002, 87, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, I.; Lee, L.H.; Ogino, S.; Marco, M.R.; Wu, C.; Chen, X.; Datta, J.; Sadot, E.; Szeglin, B.; Guillem, J.; et al. SMAD4 loss in colorectal cancer patients correlates with recurrence, loss of immune infiltrate, and chemoresistance. Clin. Cancer Res. 2018, 25, 1948–1956. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, B.; Chen, X.; Bae, S.; Singh, K.; Washington, M.K.; Datta, P.K. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br. J. Cancer 2014, 110, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, M.; Jones, E.Y. ZNRF3/RNF43 - A direct linkage of extracellular recognition and E3 ligase activity to modulate cell surface signalling. Prog. Biophys. Mol. Biol. 2015, 118, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Tsukiyama, T.; Fukui, A.; Terai, S.; Fujioka, Y.; Shinada, K.; Takahashi, H.; Yamaguchi, T.P.; Ohba, Y.; Hatakeyama, S. Molecular Role of RNF43 in Canonical and Noncanonical Wnt Signaling. Mol. Cell Biol. 2015, 35, 2007–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; Chetty, R. Rnf43. J. Clin. Pathol. 2018, 71, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loregger, A.; Grandl, M.; Mejías-Luque, R.; Allgäuer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated β-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015, 8, ra90. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.-X.; Jiang, X.; Cong, F. Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers 2016, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, C.E.; McKeone, D.M.; Kalimutho, M.; Bettington, M.L.; Pearson, S.-A.; Dumenil, T.D.; Wockner, L.F.; Burge, M.; Leggett, B.A.; Whitehall, V.L.J. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016, 7, 70589–70600. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.N.; Lai, J.C.W.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.Y.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017, 66, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl Acad Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [Green Version]
- Bagheri, M.; Tabatabae Far, M.A.; Mirzaei, H.; Ghasemi, F. Evaluation of antitumor effects of aspirin and LGK974 drugs on cellular signaling pathways, cell cycle and apoptosis in colorectal cancer cell lines compared to oxaliplatin drug. Fundam Clin. Pharm. 2020, 34, 51–64. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Neilson, K.M.; Friesel, R. Ligand-independent activation of fibroblast growth factor receptors by point mutations in the extracellular, transmembrane, and kinase domains. J. Biol. Chem. 1996, 271, 25049–25057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortes, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, K.E.; Marshall, M.E.; Heasley, L.R.; Marek, L.; Hinz, T.K.; Hercule, P.; Helfrich, B.A.; Doebele, R.C.; Heasley, L.E. Rapidly Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in NSCLC Cell Lines through De-Repression of FGFR2 and FGFR3 Expression. PLoS ONE 2010, 5, e14117. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Cufí, S.; Queralt, B.; Vazquez-Martin, A.; Martin-Castillo, B.; De Llorens, R.; Bosch-Barrera, J.; Brunet, J.; Menendez, J.A. Cross-suppression of EGFR ligands amphiregulin and epiregulin and de-repression of FGFR3 signalling contribute to cetuximab resistance in wild-type KRAS tumour cells. Br. J. Cancer 2012, 106, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [Green Version]
- Turkington, R.C.C.; Longley, D.B.B.; Allen, W.L.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.D.; Blayney, J.K.K.; Salto-Tellez, M.; Van Schaeybroeck, S.; Johnston, P.G.G.; et al. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Chen, G.; Martinka, M.; Ho, V.; Li, G. Prognostic significance of Fbw7 in human melanoma and its role in cell migration. J. Investig. Derm. 2013, 133, 1794–1802. [Google Scholar] [CrossRef] [Green Version]
- Sailo, B.L.; Banik, K.; Girisa, S.; Bordoloi, D.; Fan, L.; Halim, C.E.; Wang, H.; Kumar, A.P.; Zheng, D.; Mao, X.; et al. FBXW7 in cancer: What has been unraveled thus far? Cancers 2019, 11, 246. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Akhoondi, S.; Sun, D.; Von Der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dofou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatsuki, M.; Mimori, K.; Lshii, H.; Yokobori, T.; Takatsuno, Y.; Sato, T.; Toh, H.; Onoyama, I.; Nakayama, K.I.; Baba, H.; et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: Clinical significance. Int J. Cancer 2010, 126, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Minella, A.C.; Clurman, B.E. Mechanisms of tumor suppression by the SCFFbw7. Cell Cycle 2005, 4, 1356–1359. [Google Scholar] [CrossRef] [Green Version]
- Akhoondi, S.; Lindström, L.; Widschwendter, M.; Corcoran, M.; Bergh, J.; Spruck, C.; Grandér, D.; Sangfelt, O. Inactivation of FBXW7/hCDC4-β expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010, 12, R105. [Google Scholar] [CrossRef] [Green Version]
- Jungang, Z.; Jun, T.; Wanfu, M.; Kaiming, R. FBXW7-mediated degradation of CCDC6 is impaired by ATM during DNA damage response in lung cancer cells. Febs Lett. 2012, 586, 4257–4263. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [CrossRef] [Green Version]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Kogita, A.; Yoshioka, Y.; Sakai, K.; Togashi, Y.; Sogabe, S.; Nakai, T.; Okuno, K.; Nishio, K. Inter- and intra-tumor profiling of multi-regional colon cancer and metastasis. Biochem. Biophys. Res. Commun. 2015, 458, 52–56. [Google Scholar] [CrossRef]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etique, N.; Verzeaux, L.; Dedieu, S.; Emonard, H. Lrp-1: A checkpoint for the extracellular matrix proteolysis. BioMed Res. Int. 2013, 2013, 152163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, P.; Liao, Z.; Ren, Z.; Zhao, J.; Song, F.; Wang, G.; Chen, K.; Yang, J. Roles of low-density lipoprotein receptor-related protein 1 in tumors. Chin. J. Cancer 2016, 35, 6. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Chen, G.; Zhang, X.; Wang, Z.; Thomas, D.G.; Giordano, T.J.; Beer, D.G.; Wang, M.M. Stromal LRP1 in lung adenocarcinoma predicts clinical outcome. Clin. Cancer Res. 2011, 17, 2426–2433. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Y.; Shi, G.M.; Devbhandari, R.P.; Ke, A.W.; Wang, Y.; Wang, X.Y.; Wang, Z.; Shi, Y.H.; Xiao, Y.S.; Ding, Z.B.; et al. Low level of Low-density lipoprotein receptor-related protein 1 predicts an unfavorable prognosis of hepatocellular carcinoma after curative resection. PLoS ONE 2012, 7, e32775. [Google Scholar] [CrossRef] [Green Version]
- Baum, L.; Dong, Z.Y.; Choy, K.W.; Pang, C.P.; Ng, H.K. Low density lipoprotein receptor related protein gene amplification and 766T polymorphism in astrocytomas. Neurosci. Lett. 1998, 256, 5–8. [Google Scholar] [CrossRef]
- Catasús, L.; Llorente-Cortés, V.; Cuatrecasas, M.; Pons, C.; Espinosa, I.; Prat, J. Low-density lipoprotein receptor-related protein 1 (LRP-1) is associated with highgrade, advanced stage and p53 and p16 alterations in endometrial carcinomas. Histopathology 2011, 59, 567–571. [Google Scholar] [CrossRef]
- Catasus, L.; Gallardo, A.; Llorente-Cortes, V.; Escuin, D.; Muñoz, J.; Tibau, A.; Peiro, G.; Barnadas, A.; Lerma, E. Low-density lipoprotein receptor-related protein 1 is associated with proliferation and invasiveness in Her-2/neu and triple-negative breast carcinomas. Hum. Pathol. 2011, 42, 1581–1588. [Google Scholar] [CrossRef]
- Beneš, P.; Jurajda, M.; Žaloudík, J.; Izakovičová-Hollá, L.; Vácha, J. C766T low-density lipoprotein receptor-related protein 1 (LRP1) gene polymorphism and susceptibility to breast cancer. Breast Cancer Res. 2003, 5, R77–R81. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Li, Y.; Lee, J.; Schwartz, A.L.; Bu, G. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 2009, 69, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appert-Collin, A.; Bennasroune, A.; Jeannesson, P.; Terryn, C.; Fuhrmann, G.; Morjani, H.; Dedieu, S. Role of LRP-1 in cancer cell migration in 3-dimensional collagen matrix. Cell Adh. Migr. 2017, 11, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, P.; Gao, C.; Chen, J.; Li, J.; Chen, Z.; Xu, M.; Shao, J.; Zhang, Y.; Xie, J. Down-regulation of LRP1B in colon cancer promoted the growth and migration of cancer cells. Exp. Cell Res. 2017, 357, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.L.; Davis, A.; Gao, R.; Casasent, A.; Wang, Y.; Sei, E.; Vilar, E.; Maru, D.; Kopetz, S.; Navin, N.E. Single-cell DNA sequencing reveals a latedissemination model in metastatic colorectal cancer. Genome Res. 2017, 27, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulska, M.; Olesinski, T.; Goryca, K.; Paczkowska, K.; Statkiewicz, M.; Kopczynski, M.; Grochowska, A.; Unrug-Bielawska, K.; Tyl-Bielicka, A.; Gajewska, M.; et al. Challenges in Stratifying the Molecular Variability of Patient-Derived Colon Tumor Xenografts. BioMed Res. Int. 2018, 2018, 2954208. [Google Scholar] [CrossRef] [Green Version]
- Wolff, R.K.; Hoffman, M.D.; Wolff, E.C.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.S.; Slattery, M.L. Mutation analysis of adenomas and carcinomas of the colon: Early and late drivers. Genes Chromosomes Cancer 2018, 57, 366–376. [Google Scholar] [CrossRef]
- Wang, X.; Nagl, N.G.; Wilsker, D.; Van Scoy, M.; Pacchione, S.; Yaciuk, P.; Dallas, P.B.; Moran, E. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem. J. 2004, 383, 319–325. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- Toumpeki, C.; Liberis, A.; Tsirkas, I.; Tsirka, T.; Kalagasidou, S.; Inagamova, L.; Anthoulaki, X.; Tsatsaris, G.; Kontomanolis, E.N. The Role of ARID1A in Endometrial Cancer and the Molecular Pathways Associated With Pathogenesis and Cancer Progression. In Vivo 2019, 33, 659–667. [Google Scholar] [CrossRef]
- Mariotti, V.; McLeod, H.L.; Soliman, H.H. ARID1a as a marker of prognosis and increased sensitivity to CDK4/6, mTOR 1/2 and Src homology region 2 phosphatase (SHP 1/2) inhibitors in breast cancer (BC). J. Clin. Oncol. 2019, 37, 1082. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachaliou, N.; Bracht, J.W.P.; Rosell, R. ARID1A Gene Driver Mutations in Lung Adenocarcinomas. J. Thorac. Oncol. 2018, 13, e255–e257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-S.; Jeong, H.; Choi, J.-W.; Oh, H.E.; Lee, J.-H. Unique characteristics of ARID1A mutation and protein level in gastric and colorectal cancer: A meta-analysis. Saudi J. Gastroenterol. 2017, 23, 268–274. [Google Scholar] [PubMed]
- Cajuso, T.; Hänninen, U.A.; Kondelin, J.; Gylfe, A.E.; Tanskanen, T.; Katainen, R.; Pitkänen, E.; Ristolainen, H.; Kaasinen, E.; Taipale, M.; et al. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int. J. Cancer 2014, 135, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Nakayama, K.; Razia, S.; Nakamura, K.; Ishikawa, M.; Minamoto, T.; Ishibashi, T.; Yamashita, H.; Iida, K.; Kyo, S. ARID1B as a Potential Therapeutic Target for ARID1A-Mutant Ovarian Clear Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 1710. [Google Scholar] [CrossRef] [Green Version]
- Sen, M.; Wang, X.; Hamdan, F.H.; Rapp, J.; Eggert, J.; Kosinsky, R.L.; Wegwitz, F.; Kutschat, A.P.; Younesi, F.S.; Gaedcke, J.; et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin. Epigenetics 2019, 11, 92. [Google Scholar] [CrossRef]
- Thomas, R.K.; Baker, A.C.; DeBiasi, R.M.; Winckler, W.; LaFramboise, T.; Lin, W.M.; Wang, M.; Feng, W.; Zander, T.; MacConaill, L.E.; et al. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 2007, 39, 347–351. [Google Scholar] [CrossRef]
- Macintyre, G.; Ylstra, B.; Brenton, J.D. Sequencing Structural Variants in Cancer for Precision Therapeutics. Trends Genet. 2016, 32, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, F.; Charbonnier, F.; Martin, C.; Frerot, S.; Olschwang, S.; Wang, Q.; Boisson, C.; Buisine, M.P.; Nilbert, M.; Lindblom, A.; et al. Screening for genomic rearrangements of the MMR genes must be included in the routine diagnosis of HNPCC. J. Med. Genet. 2004, 41, 18–20. [Google Scholar] [CrossRef] [Green Version]
- Van Der Klift, H.; Wijnen, J.; Wagner, A.; Verkuilen, P.; Tops, C.; Otway, R.; Kohonen-Corish, M.; Vasen, H.; Oliani, C.; Barana, D.; et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 2005, 44, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Cavallo, A.; Liccardo, R.; Cudia, B.; De Rosa, M.; Diana, G.; Izzo, P. Contribution of large genomic rearrangements in Italian Lynch syndrome patients: Characterization of a novel alu-mediated deletion. BioMed Res. Int. 2013, 2013, 219897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Kwak, Y.; Nam, K.H.; Kim, D.-W.; Kang, S.-B.; Choe, G.; Kim, W.H.; Lee, H.S. c-MYC Copy-Number Gain Is an Independent Prognostic Factor in Patients with Colorectal Cancer. PLoS ONE 2015, 10, e0139727. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Hatakeyama, K.; Nagashima, T.; Watanabe, Y.; Kanto, K.; Doi, Y.; Ide, T.; Shimoda, Y.; Tanabe, T.; Ohnami, S.; et al. Integrated analysis of gene expression and copy number identified potential cancer driver genes with amplification-dependent overexpression in 1,454 solid tumors. Sci. Rep. 2017, 7, 641. [Google Scholar] [CrossRef]
- He, W.-L.; Weng, X.-T.; Wang, J.-L.; Lin, Y.-K.; Liu, T.-W.; Zhou, Q.-Y.; Hu, Y.; Pan, Y.; Chen, X.-L. Association Between c-Myc and Colorectal Cancer Prognosis: A Meta-Analysis. Front. Physiol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Kwak, Y.; Yun, S.; Nam, S.K.; Seo, A.N.; Lee, K.S.; Shin, E.; Oh, H.-K.; Kim, D.W.; Kang, S.B.; Kim, W.H.; et al. Comparative analysis of the EGFR, HER2, c-MYC, and MET variations in colorectal cancer determined by three different measures: Gene copy number gain, amplification status and the 2013 ASCO/CAP guideline criterion for HER2 testing of breast cancer. J. Transl. Med. 2017, 15, 167. [Google Scholar] [CrossRef] [Green Version]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Sakurai, M.; Enjoji, S.; Kawasaki, H.; Umata, K.; Ohama, T.; Fujiwara, N.; Yabe, R.; Tsuji, S.; Yamawaki, H.; et al. Establishment of a Novel Model for Anticancer Drug Resistance in Three-Dimensional Primary Culture of Tumor Microenvironment. Stem Cells Int. 2016, 2016, 7053872. [Google Scholar] [CrossRef]

{kind=link}
| Altered Gene | Prevalence in CRC | Actionable and/or Predictive Value | Highest Level of Evidence | In vitro or In vivo Investigation in CRC and/or Other Cancers |
|---|---|---|---|---|
| SMAD4 | 2%–20% [23,24] | Resistance to anti-EGFR monoclonal antibodies, cetuximab as a single agent or in combination with standard chemotherapeutic agents [25]. | Retrospective Cohorts | SMAD4 deficiency induces 5 fluorouracil (5FU) chemoresistance in CT26 and SW620 cells via the activation of PI3K/Akt/CDC2/survivin pathway. The PI3K inhibitor, LY294002, able to trigger 5FU chemosensitivity via cell cycle arrest by hindering the PI3K/Akt/CDC2/survivin cascade in the SMAD4-deficient cells [27]. |
| Unresponsive to anti-epidermal growth receptor therapy and significantly shorter-progression-free survival durations [26]. | Retrospective Cohorts | |||
| RNF43 | 6%–18% [28,29] | Sensitive to LGK974 for pancreatic cell line with RNF43 loss of function mutation [30]. | Case Study | RNF43 knockdown enhances the tumorigenic potential of CRC cell lines in vitro and in vivo. Larger tumors were observed in the RNF43 knockout mouse model [32]. |
| Phase I evaluation of LGK974 in melanoma, breast cancer (lobular or triple-negative) and pancreatic cancer [31]. | Phase I Clinical Trial | |||
| FGFRs | None was reported in one CRC study [33]; however, TCGA studies reported 1.7%–5% of CRC patients harbored alteration in FGFR genes [34] | Sensitive to FGFR Tyrosine Kinase Inhibitor (TKIs), AZD4547, as reported by Phase I and II clinical trials in gastric cancers [36]. | Phase II Clinical Trial | FGFR2 amplification and overexpression were implicated in survival and proliferation of CRC cell line NCI-H716 and sensitive to FGFR inhibitors [37]. |
| In other cancers: FGFR1: 3.5% FGFR2: 1.5% FGFR3: 2.0% FGFR4: 0.5% [35] | FGFR tyrosine-kinase inhibitors (TKIs), AZD4547, demonstrated growth inhibition in the colorectal cell line with FGFR2 amplification [37]. | Preclinical | ||
| FBXW7 | 6%–20% [20,38,39,40,41] | Sensitive to mTOR inhibitors rapamycin in breast cancer cell line with the loss of FBXW7 and deletion or mutation of PTEN [42]. | Preclinical | Mutated CRC cell lines are less sensitive to regorafenib and sorafenib [45]. |
| Better clinical outcome in T-cell acute lymphoblastic leukaemia (T-ALL) patients [43]. | Clinical | |||
| mCRC patients harboring FBXW7 missense mutations had significantly worse overall survival than those with wild-type FBXW7 [44]. | Retrospective Cohorts | |||
| LRP1 | 6% [23,46] | mCRC patients with mutations and low expression of LRP1 had poor clinical outcomes even though after treatment with bevacizumab [46]. | Retrospective Cohorts | LRP1 together with its ligands, tissue plasminogen activator (tPA), regulate melanoma growth and lung metastasis in vivo [47]. |
| ARID1A | 6.2%–10.9% [34,48] | ARID1A protein loss, due to mutations, is associated with the late TNM stage, distant metastasis, and poor pathologic differentiation in CRC patients [49] | Retrospective Cohorts | ARID1A overexpression in SW620 cell line inhibits proliferation and facilitated 5-FU-induced apoptosis. ARID1A knockdown in SW480 cell line promotes proliferation and inhibited 5-FU-induced apoptosis [50]. |
| Stage IV patients with ARID1A protein loss in primary tumors had longer survival compared to those with ARID1A positive tumors [49] | CRC cell lines with mutated ARID1A are are selectively sensitized to ionizing radiation after knockdown of its other subunit, ARID1B [51]. |
| Gene A | Gene B | Log2 Odds Ratio | q-Value | Tendency |
|---|---|---|---|---|
| ARID1A | FGFR3 | >3 | <0.001 | Co-occurrence |
| RNF43 | FGFR2 | >3 | <0.001 | Co-occurrence |
| RNF43 | FGFR3 | >3 | <0.001 | Co-occurrence |
| LRP1 | FGFR2 | >3 | <0.001 | Co-occurrence |
| ARID1A | FGFR2 | 2.99 | <0.001 | Co-occurrence |
| FGFR2 | FGFR1 | 2.784 | <0.001 | Co-occurrence |
| RNF43 | LRP1 | 2.618 | <0.001 | Co-occurrence |
| FGFR3 | FGFR4 | 2.6 | <0.001 | Co-occurrence |
| FGFR2 | FGFR4 | 2.532 | <0.001 | Co-occurrence |
| ARID1A | RNF43 | 2.503 | <0.001 | Co-occurrence |
| FGFR2 | FGFR3 | 2.413 | 0.001 | Co-occurrence |
| LRP1 | FGFR3 | 2.411 | <0.001 | Co-occurrence |
| RNF43 | FGFR4 | 2.344 | <0.001 | Co-occurrence |
| LRP1B | FGFR3 | 2.339 | <0.001 | Co-occurrence |
| ARID1A | LRP1 | 2.202 | <0.001 | Co-occurrence |
| FGFR1 | FGFR3 | 2.02 | <0.001 | Co-occurrence |
| FGFR1 | FGFR4 | 1.974 | 0.001 | Co-occurrence |
| FBXW7 | FGFR3 | 1.926 | <0.001 | Co-occurrence |
| FBXW7 | FGFR2 | 1.913 | <0.001 | Co-occurrence |
| LRP1 | FGFR4 | 1.737 | 0.004 | Co-occurrence |
| LRP1 | LRP1B | 1.651 | <0.001 | Co-occurrence |
| FBXW7 | LRP1 | 1.568 | <0.001 | Co-occurrence |
| RNF43 | FGFR1 | 1.459 | <0.001 | Co-occurrence |
| ARID1A | LRP1B | 1.447 | <0.001 | Co-occurrence |
| LRP1B | FGFR2 | 1.41 | 0.005 | Co-occurrence |
| FBXW7 | FGFR4 | 1.318 | 0.002 | Co-occurrence |
| LRP1 | FGFR1 | 1.316 | 0.005 | Co-occurrence |
| LRP1B | FGFR4 | 1.247 | 0.016 | Co-occurrence |
| ARID1A | FBXW7 | 1.216 | <0.001 | Co-occurrence |
| RNF43 | LRP1B | 1.186 | <0.001 | Co-occurrence |
| LRP1B | FGFR1 | 1.121 | 0.004 | Co-occurrence |
| FBXW7 | RNF43 | 1.111 | <0.001 | Co-occurrence |
| ARID1A | FGFR4 | 1.031 | 0.032 | Co-occurrence |
| ARID1A | FGFR1 | 0.988 | 0.004 | Co-occurrence |
| FGFR4 | SMAD4 | 0.969 | 0.018 | Co-occurrence |
| FBXW7 | LRP1B | 0.905 | <0.001 | Co-occurrence |
| FGFR3 | SMAD4 | 0.824 | 0.036 | Co-occurrence |
| FGFR1 | SMAD4 | 0.726 | 0.01 | Co-occurrence |
| FBXW7 | FGFR1 | 0.691 | 0.016 | Co-occurrence |
| LRP1B | SMAD4 | 0.637 | 0.006 | Co-occurrence |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Yunos, R.I.; Ab Mutalib, N.S.; Tieng, F.Y.F.; Abu, N.; Jamal, R. Actionable Potentials of Less Frequently Mutated Genes in Colorectal Cancer and Their Roles in Precision Medicine. Biomolecules 2020, 10, 476. https://doi.org/10.3390/biom10030476
Mohd Yunos RI, Ab Mutalib NS, Tieng FYF, Abu N, Jamal R. Actionable Potentials of Less Frequently Mutated Genes in Colorectal Cancer and Their Roles in Precision Medicine. Biomolecules. 2020; 10(3):476. https://doi.org/10.3390/biom10030476
Chicago/Turabian StyleMohd Yunos, Ryia Illani, Nurul Syakima Ab Mutalib, Francis Yew Fu Tieng, Nadiah Abu, and Rahman Jamal. 2020. "Actionable Potentials of Less Frequently Mutated Genes in Colorectal Cancer and Their Roles in Precision Medicine" Biomolecules 10, no. 3: 476. https://doi.org/10.3390/biom10030476