TGF-β Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
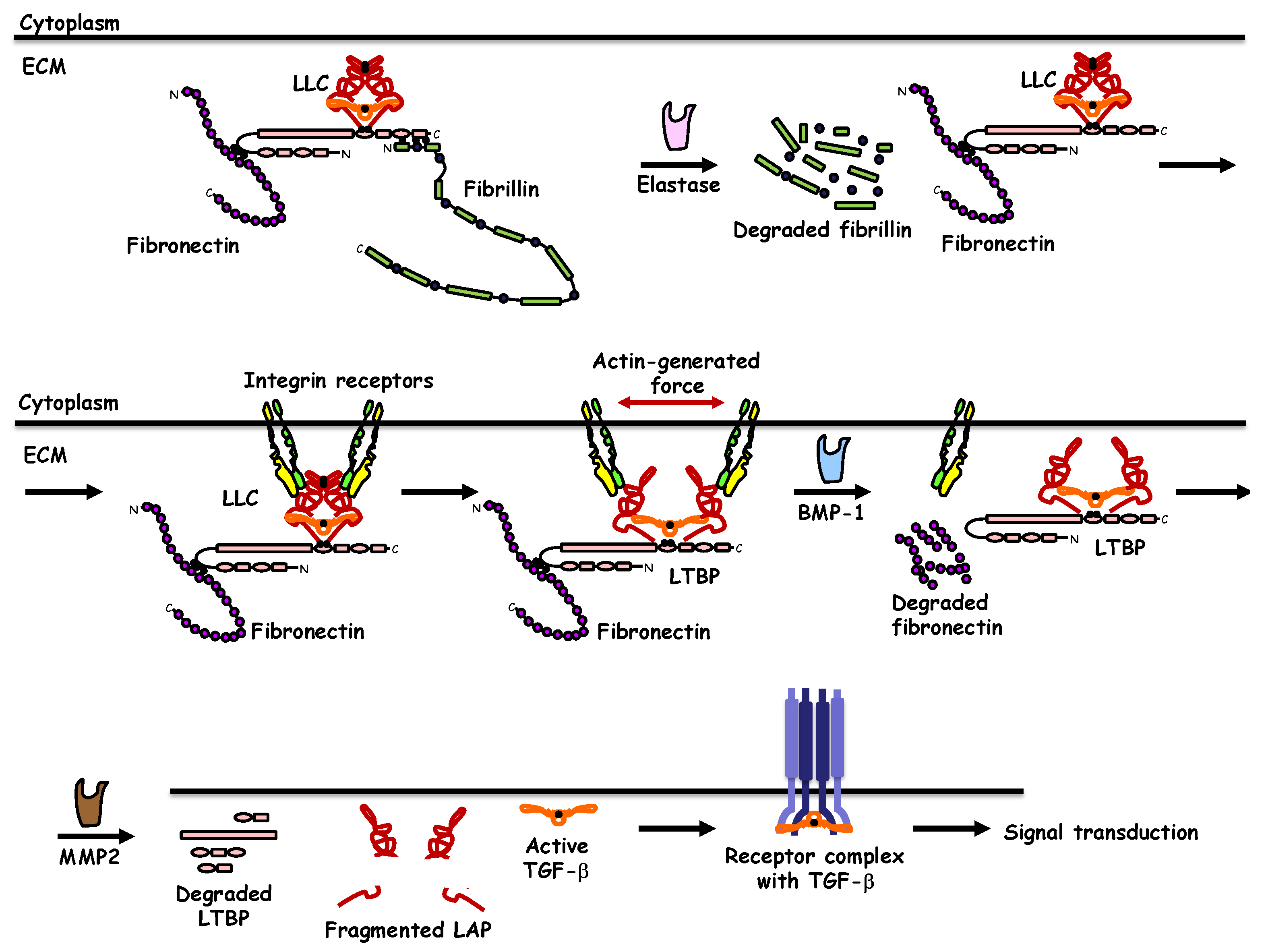
2. TGF-β Synthesis, Extracellular Deposition, and Activation
3. Receptors for TGF-β Family Members
4. Coreceptors Facilitate or Inhibit Signaling via the TGF-β Receptors
5. TGF-β Receptor Internalization, Degradation, and Recycling
6. SMAD Signaling
6.1. Structure of SMADs
6.2. Activation of SMADs
6.3. Nucleocytoplasmic Shuttling of SMADs
6.4. Posttranslational Modifications of SMADs
6.5. SMADs in Transcription
6.6. SMADs in Posttranscriptional Regulation
6.7. Function and Regulation of Inhibitory SMADs
7. Non-SMAD TGF-β Signaling Pathways
7.1. ERK MAP Kinase Pathway
7.2. JNK and p38 MAP Kinase Pathways (via TAK1)
7.3. PI3K-AKT Pathway
7.4. TGF-β Type I Receptor Intracellular Domain Signaling
7.5. JAK-STAT Pathway
7.6. Rho-(like) GTPase Pathway
7.7. Other TGF-β Activated SMAD-Independent Pathways
8. Signaling Cross-Talk
8.1. Cross-Talk at the Receptor Level
8.2. Cross-Talk at the Level of SMAD (or Other Signaling Effector) Activation
9. Future Perspectives and Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Heldin, C.H.; Lu, B.; Evans, R.; Gutkind, J.S. Signals and Receptors. Cold Spring Harb. Perspect. Biol. 2016, 8, a005900. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Miyazono, K. The TGF-β Family; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2008; pp. 1–1114. [Google Scholar]
- Huminiecki, L.; Goldovsky, L.; Freilich, S.; Moustakas, A.; Ouzounis, C.A.; Heldin, C.H. Emergence, development and diversification of the TGF-β signalling pathway within the animal kingdom. BMC Evol. Biol. 2009, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Heldin, C.H. The regulation of TGFβ signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFb signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Hill, C.S. Beyond TGFβ: Roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Massagué, J. Publisher Correction: Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 479. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Heldin, C.H. Role for carbohydrate structures in TGF-β1 latency. Nature 1989, 338, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Liénart, S.; Merceron, R.; Vanderaa, C.; Lambert, F.; Colau, D.; Stockis, J.; van der Woning, B.; De Haard, H.; Saunders, M.; Coulie, P.G.; et al. Structural basis of latent TGF-β1 presentation and activation by GARP on human regulatory T cells. Science 2018, 362, 952–956. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Garrison, B.S.; Ma, W.; Wang, R.; Jiang, A.; Li, J.; Mistry, M.; Bronson, R.T.; Santoro, D.; Franco, C.; et al. A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System. Cell 2018, 174, 156–171. [Google Scholar] [CrossRef] [Green Version]
- Fuerer, C.; Nostro, M.C.; Constam, D.B. Nodal.Gdf1 heterodimers with bound prodomains enable serum-independent nodal signaling and endoderm differentiation. J. Biol. Chem. 2014, 289, 17854–17871. [Google Scholar] [CrossRef] [Green Version]
- Little, S.C.; Mullins, M.C. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis. Nat. Cell Biol. 2009, 11, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Ge, G.; Greenspan, D.S. BMP1 controls TGFβ1 activation via cleavage of latent TGFβ-binding protein. J. Cell Biol. 2006, 175, 111–120. [Google Scholar] [CrossRef]
- Ge, G.; Greenspan, D.S. Developmental roles of the BMP1/TLD metalloproteinases. Birth Defects Res. Part C Embryo Today 2006, 78, 47–68. [Google Scholar] [CrossRef]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin αvβ6 binds and activates latent TGFβ1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of TGF-β. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 2008, 29, 157–168. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Martin, J.; Manganaro, T.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGFβ family type I receptors. Cell 1996, 86, 435–444. [Google Scholar]
- Huse, M.; Chen, Y.G.; Massagué, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF β receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Mulder, K.M.; Morris, S.L. Activation of p21ras by transforming growth factor β in epithelial cells. J. Biol. Chem. 1992, 267, 5029–5031. [Google Scholar]
- Hartsough, M.T.; Mulder, K.M. Transforming growth factor β activation of p44mapk in proliferating cultures of epithelial cells. J. Biol. Chem. 1995, 270, 7117–7124. [Google Scholar]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, L.; Wells, R.G.; Lodish, H.F.; Henis, Y.I. Oligomeric structure of type I and type II transforming growth factor β receptors: Homodimers form in the ER and persist at the plasma membrane. J. Cell Biol. 1998, 140, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Gutman, O.; Knaus, P.; Henis, Y.I. Oligomeric interactions of TGF-β and BMP receptors. FEBS Lett. 2012, 586, 1885–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rechtman, M.M.; Nakaryakov, A.; Shapira, K.E.; Ehrlich, M.; Henis, Y.I. Different domains regulate homomeric and heteromeric complex formation among type I and type II transforming growth factor-β receptors. J. Biol. Chem. 2009, 284, 7843–7852. [Google Scholar] [CrossRef] [Green Version]
- Radaev, S.; Zou, Z.C.; Huang, T.; Lafer, E.M.; Hinck, A.P.; Sun, P.D. Ternary Complex of Transforming Growth Factor-β 1 Reveals Isoform-specific Ligand Recognition and Receptor Recruitment in the Superfamily. J. Biol. Chem. 2010, 285, 14806–14814. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massagué, J. The TGF β receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Huang, T.; David, L.; Mendoza, V.; Yang, Y.; Villarreal, M.; De, K.; Sun, L.; Fang, X.; Lopez-Casillas, F.; Wrana, J.L.; et al. TGF-β signalling is mediated by two autonomously functioning TβRI:TβRII pairs. EMBO J. 2011, 30, 1263–1276. [Google Scholar] [CrossRef] [Green Version]
- López-Casillas, F.; Cheifetz, S.; Doody, J.; Andres, J.L.; Lane, W.S.; Massagué, J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell 1991, 67, 785–795. [Google Scholar] [CrossRef]
- Wang, X.F.; Lin, H.Y.; Ng-Eaton, E.; Downward, J.; Lodish, H.F.; Weinberg, R.A. Expression cloning and characterization of the TGF-β type III receptor. Cell 1991, 67, 797–805. [Google Scholar] [CrossRef]
- Nickel, J.; ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [Green Version]
- López-Casillas, F.; Payne, H.M.; Andres, J.L.; Massagué, J. Betaglycan can act as a dual modulator of TGF-β access to signaling receptors: Mapping of ligand binding and GAG attachment sites. J. Cell Biol. 1994, 124, 557–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Lin, H.Y.; Henis, Y.I.; Plamondon, J.; O’Connor-McCourt, M.D.; Lodish, H.F. The transforming growth factor β receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem. 1993, 268, 22215–22218. [Google Scholar]
- Lin, H.Y.; Moustakas, A.; Knaus, P.; Wells, R.G.; Henis, Y.I.; Lodish, H.F. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-β receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-β ligands. J. Biol. Chem. 1995, 270, 2747–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenvers, K.L.; Tursky, M.L.; Harder, K.W.; Kountouri, N.; Amatayakul-Chantler, S.; Grail, D.; Small, C.; Weinberg, R.A.; Sizeland, A.M.; Zhu, H.J. Heart and liver defects and reduced transforming growth factor β2 sensitivity in transforming growth factor β type III receptor-deficient embryos. Mol. Cell. Biol. 2003, 23, 4371–4385. [Google Scholar] [PubMed] [Green Version]
- You, H.J.; Bruinsma, M.W.; How, T.; Ostrander, J.H.; Blobe, G.C. The type III TGF-β receptor signals through both Smad3 and the p38 MAP kinase pathways to contribute to inhibition of cell proliferation. Carcinogenesis 2007, 28, 2491–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; How, T.; Kirkbride, K.C.; Gordon, K.J.; Lee, J.D.; Hempel, N.; Kelly, P.; Moeller, B.J.; Marks, J.R.; Blobe, G.C. The type III TGF-β receptor suppresses breast cancer progression. J. Clin. Investig. 2007, 117, 206–217. [Google Scholar]
- Mendoza, V.; Vilchis-Landeros, M.M.; Mendoza-Hernandez, G.; Huang, T.; Villarreal, M.M.; Hinck, A.P.; Lopez-Casillas, F.; Montiel, J.L. Betaglycan has two independent domains required for high affinity TGF-β binding: Proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry 2009, 48, 11755–11765. [Google Scholar]
- Eickelberg, O.; Centrella, M.; Reiss, M.; Kashgarian, M.; Wells, R.G. Betaglycan inhibits TGF-β signaling by preventing type I-type II receptor complex formation. Glycosaminoglycan modifications alter betaglycan function. J. Biol. Chem. 2002, 277, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Wiater, E.; Harrison, C.A.; Lewis, K.A.; Gray, P.C.; Vale, W.W. Identification of distinct inhibin and transforming growth factor β-binding sites on betaglycan: Functional separation of betaglycan co-receptor actions. J. Biol. Chem. 2006, 281, 17011–17022. [Google Scholar]
- Lee, N.Y.; Kirkbride, K.C.; Sheu, R.D.; Blobe, G.C. The transforming growth factor-β type III receptor mediates distinct subcellular trafficking and downstream signaling of activin-like kinase (ALK)3 and ALK6 receptors. Mol. Biol. Cell 2009, 20, 4362–4370. [Google Scholar] [CrossRef] [Green Version]
- Knelson, E.H.; Gaviglio, A.L.; Tewari, A.K.; Armstrong, M.B.; Nixon, A.B.; Starr, M.D.; Mythreye, K.; Blobe, G.C. The type III TGF-β receptor promotes FGF2-mediated neuronal differentiation in neuroblastoma. Cancer Res. 2013, 73, 4786–4798. [Google Scholar] [CrossRef] [Green Version]
- Barbara, N.P.; Wrana, J.L.; Letarte, M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-β superfamily. J. Biol. Chem. 1999, 274, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Esteo, M.; Sanchez-Elsner, T.; Letamendia, A.; Bernabeu, C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-β receptors I and II. J. Biol. Chem. 2002, 277, 29197–29209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, B.N.; Lee, N.Y.; How, T.; Blobe, G.C. ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, S.; Alvarez-Munoz, P.; Pericacho, M.; Dijke, P.T.; Bernabeu, C.; Lopez-Novoa, J.M.; Rodriguez-Barbero, A. L- and S-endoglin differentially modulate TGFβ1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Malhotra, R.; Paskin-Flerlage, S.; Zamanian, R.T.; Zimmerman, P.; Schmidt, J.W.; Deng, D.Y.; Southwood, M.; Spencer, R.; Lai, C.S.; Parker, W.; et al. Circulating angiogenic modulatory factors predict survival and functional class in pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.T.; Liu, J.J.; Luo, Y.; Chaosu, E.; Haltiwanger, R.S.; Abate-Shen, C.; Shen, M.M. Dual roles of Cripto as a ligand and coreceptor in the nodal signaling pathway. Mol. Cell. Biol. 2002, 22, 4439–4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, P.C.; Shani, G.; Aung, K.; Kelber, J.; Vale, W. Cripto binds transforming growth factor β (TGF-β) and inhibits TGF-β signaling. Mol. Cell. Biol. 2006, 26, 9268–9278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnson, K.W.; Tam, B.Y.; Liu, K.; Marcoux, A.; Lepage, P.; Roy, S.; Bizet, A.A.; Philip, A. Identification of CD109 as part of the TGF-β receptor system in human keratinocytes. FASEB J. 2006, 20, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Dellus, H.; Massagué, J.; Niehrs, C. Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, T.; Oda, T.; Matsuura, K.; Akiyama, T. Transcriptional regulation of the TGF-β pseudoreceptor BAMBI by TGF-β signaling. Biochem. Biophys. Res. Commun. 2004, 320, 680–684. [Google Scholar] [CrossRef]
- Yan, X.; Lin, Z.; Chen, F.; Zhao, X.; Chen, H.; Ning, Y.; Chen, Y.G. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-β signaling. J. Biol. Chem. 2009, 284, 30097–30104. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; de Boeck, M.; van Dam, H.; ten Dijke, P. Regulation of the TGF-β pathway by deubiquitinases in cancer. Int. J. Biochem. Cell Biol. 2016, 76, 135–145. [Google Scholar] [CrossRef]
- Doré, J.J., Jr.; Yao, D.; Edens, M.; Garamszegi, N.; Sholl, E.L.; Leof, E.B. Mechanisms of transforming growth factor-β receptor endocytosis and intracellular sorting differ between fibroblasts and epithelial cells. Mol. Biol. Cell 2001, 12, 675–684. [Google Scholar] [CrossRef]
- Murphy, S.J.; Dore, J.J.; Edens, M.; Coffey, R.J.; Barnard, J.A.; Mitchell, H.; Wilkes, M.; Leof, E.B. Differential trafficking of transforming growth factor-β receptors and ligand in polarized epithelial cells. Mol. Biol. Cell 2004, 15, 2853–2862. [Google Scholar] [CrossRef]
- Barrios-Rodiles, M.; Brown, K.R.; Ozdamar, B.; Bose, R.; Liu, Z.; Donovan, R.S.; Shinjo, F.; Liu, Y.; Dembowy, J.; Taylor, I.W.; et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005, 307, 1621–1625. [Google Scholar] [CrossRef] [Green Version]
- Hayes, S.; Chawla, A.; Corvera, S. TGF β receptor internalization into EEA1-enriched early endosomes: Role in signaling to Smad2. J. Cell Biol. 2002, 158, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Penheiter, S.G.; Mitchell, H.; Garamszegi, N.; Edens, M.; Dore, J.J., Jr.; Leof, E.B. Internalization-dependent and -independent requirements for transforming growth factor β receptor signaling via the Smad pathway. Mol. Cell. Biol. 2002, 22, 4750–4759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Böttinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (TGF)-β/SMAD signaling through an interaction with the TGF-β type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [Green Version]
- Bizet, A.A.; Liu, K.; Tran-Khanh, N.; Saksena, A.; Vorstenbosch, J.; Finnson, K.W.; Buschmann, M.D.; Philip, A. The TGF-β co-receptor, CD109, promotes internalization and degradation of TGF-β receptors. Biochim. Biophys. Acta 2011, 1813, 742–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizet, A.A.; Tran-Khanh, N.; Saksena, A.; Liu, K.; Buschmann, M.D.; Philip, A. CD109-mediated degradation of TGF-β receptors and inhibition of TGF-β responses involve regulation of SMAD7 and Smurf2 localization and function. J. Cell. Biochem. 2012, 113, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.L.; et al. c-Cbl-Mediated Neddylation Antagonizes Ubiquitination and Degradation of the TGF-β Type II Receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atfi, A.; Dumont, E.; Colland, F.; Bonnier, D.; L’Helgoualc’h, A.; Prunier, C.; Ferrand, N.; Clément, B.; Wewer, U.M.; Théret, N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-β signaling through interaction with the type II receptor. J. Cell Biol. 2007, 178, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Kirkbride, K.C.; How, T.; Nelson, C.D.; Mo, J.; Frederick, J.P.; Wang, X.F.; Lefkowitz, R.J.; Blobe, G.C. β-Arrestin 2 mediates endocytosis of type III TGF-β receptor and down-regulation of its signaling. Science 2003, 301, 1394–1397. [Google Scholar] [CrossRef]
- Finger, E.C.; Lee, N.Y.; You, H.J.; Blobe, G.C. Endocytosis of the type III transforming growth factor-β (TGF-β) receptor through the clathrin-independent/lipid raft pathway regulates TGF-β signaling and receptor down-regulation. J. Biol. Chem. 2008, 283, 34808–34818. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, H.; Choudhury, A.; Pagano, R.E.; Leof, E.B. Ligand-dependent and -independent transforming growth factor-β receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol. Biol. Cell 2004, 15, 4166–4178. [Google Scholar] [CrossRef] [PubMed]
- Penheiter, S.G.; Singh, R.D.; Repellin, C.E.; Wilkes, M.C.; Edens, M.; Howe, P.H.; Pagano, R.E.; Leof, E.B. Type II Transforming Growth Factor-β Receptor Recycling Is Dependent upon the Clathrin Adaptor Protein Dab2. Mol. Biol. Cell 2010, 21, 4009–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.Q.; Murphy, S.J.; Wilkes, M.C.; Ji, Y.; Leof, E.B. Retromer maintains basolateral distribution of the type II TGF-β receptor via the recycling endosome. Mol. Biol. Cell 2013, 24, 2285–2298. [Google Scholar] [CrossRef] [PubMed]
- Yakymovych, I.; Yakymovych, M.; Zang, G.; Mu, Y.; Bergh, A.; Landström, M.; Heldin, C.H. CIN85 modulates TGFβ signaling by promoting the presentation of TGFβ receptors on the cell surface. J. Cell Biol. 2015, 210, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Sekelsky, J.J.; Newfeld, S.J.; Raftery, L.A.; Chartoff, E.H.; Gelbart, W.M. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 1995, 139, 1347–1358. [Google Scholar] [PubMed]
- Savage, C.; Das, P.; Finelli, A.L.; Townsend, S.R.; Sun, C.Y.; Baird, S.E.; Padgett, R.W. Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor β pathway components. Proc. Natl. Acad. Sci. USA 1996, 93, 790–794. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, X.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 1996, 383, 168–172. [Google Scholar] [CrossRef]
- Piek, E.; Moustakas, A.; Kurisaki, A.; Heldin, C.H.; ten Dijke, P. TGF-β type I receptor/ALK5 and SMAD proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 1999, 112, 4557–4568. [Google Scholar]
- Shi, Y.; Wang, Y.F.; Jayaraman, L.; Yang, H.J.; Massagué, J.; Pavletich, N.P. Crystal structure of a Smad MH1 domain bound to DNA: Insights on DNA binding in TGF-β signaling. Cell 1998, 94, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.G.; Hata, A.; Lo, R.S.; Wotton, D.; Shi, Y.; Pavletich, N.; Massagué, J. Determinants of specificity in TGF-β signal transduction. Genes Dev. 1998, 12, 2144–2152. [Google Scholar] [CrossRef] [Green Version]
- Lo, R.S.; Chen, Y.G.; Shi, Y.; Pavletich, N.P.; Massagué, J. The L3 loop: A structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J. 1998, 17, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Hata, A.; Lo, R.S.; Massagué, J.; Pavletich, N.P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 1997, 388, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Sampath, K. Alternative splicing of SMADs in differentiation and tissue homeostasis. Dev. Growth Differ. 2010, 52, 335–342. [Google Scholar] [CrossRef]
- Souchelnytskyi, S.; Tamaki, K.; Engström, U.; Wernstedt, C.; ten Dijke, P.; Heldin, C.H. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J. Biol. Chem. 1997, 272, 28107–28115. [Google Scholar] [CrossRef] [Green Version]
- Persson, U.; Izumi, H.; Souchelnytskyi, S.; Itoh, S.; Grimsby, S.; Engström, U.; Heldin, C.H.; Funa, K.; ten Dijke, P. The L45 loop in type I receptors for TGF-β family members is a critical determinant in specifying Smad isoform activation. FEBS Lett. 1998, 434, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Abdollah, S.; Macías-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.K.; Bergmann, S.; Pandolfi, P.P. Cytoplasmic PML function in TGF-β signalling. Nature 2004, 431, 205–211. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Smine, A.; Xu, X.X.; Howe, P.H. The adaptor molecule Disabled-2 links the transforming growth factor β receptors to the Smad pathway. EMBO J. 2001, 20, 2789–2801. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Chen, Y.G.; Ozdamar, B.; Gyuricza, C.A.; Chong, P.A.; Wrana, J.L.; Massagué, J.; Shi, Y. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Takeshita, T.; Asao, H.; Kimura, Y.; Murata, K.; Sasaki, Y.; Hanai, J.I.; Beppu, H.; Tsukazaki, T.; Wrana, J.L.; et al. Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol. Cell. Biol. 2000, 20, 9346–9355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacko, B.M.; Qin, B.Y.; Tiwari, A.; Shi, G.; Lam, S.; Hayward, L.J.; De Caestecker, M.; Lin, K. Structural basis of heteromeric Smad protein assembly in TGF-β signaling. Mol. Cell 2004, 15, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hu, M.; Chai, J.; Seoane, J.; Huse, M.; Li, C.; Rigotti, D.J.; Kyin, S.; Muir, T.W.; Fairman, R.; et al. Crystal structure of a phosphorylated Smad2. Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-β signaling. Mol. Cell 2001, 8, 1277–1289. [Google Scholar] [CrossRef]
- Kawabata, M.; Inoue, H.; Hanyu, A.; Imamura, T.; Miyazono, K. Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J. 1998, 17, 4056–4065. [Google Scholar] [CrossRef]
- Lucarelli, P.; Schilling, M.; Kreutz, C.; Vlasov, A.; Boehm, M.; Iwamoto, N.; Steiert, B.; Lattermann, S.; Wäsch, M.; Stepath, M.; et al. Resolving the Combinatorial Complexity of Smad Protein Complex Formation and Its Link to Gene Expression. Cell Syst. 2018, 6, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.J.; Yue, J.B.; Frey, R.S.; Zhu, Q.C.; Mulder, K.M. Transforming growth factor β signaling through Smad1 in human breast cancer cells. Cancer Res. 1998, 58, 4752–4757. [Google Scholar] [PubMed]
- Ramachandran, A.; Vizan, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Muller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. Elife 2018, 7, e31756. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurisaki, A.; Kose, S.; Yoneda, Y.; Heldin, C.H.; Moustakas, A. Transforming growth factor-β induces nuclear import of Smad3 in an importin-β1 and Ran-dependent manner. Mol. Biol. Cell 2001, 12, 1079–1091. [Google Scholar] [CrossRef]
- Kawasaki, N.; Miwa, T.; Hokari, S.; Sakurai, T.; Ohmori, K.; Miyauchi, K.; Miyazono, K.; Koinuma, D. Long noncoding RNA NORAD regulates transforming growth factor-β signaling and epithelial-to-mesenchymal transition-like phenotype. Cancer Sci. 2018, 109, 2211–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Latek, R.; Lodish, H.F. An extended bipartite nuclear localization signal in Smad4 is required for its nuclear import and transcriptional activity. Oncogene 2003, 22, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurisaki, A.; Kurisaki, K.; Kowanetz, M.; Sugino, H.; Yoneda, Y.; Heldin, C.H.; Moustakas, A. The mechanism of nuclear export of Smad3 involves exportin 4 and Ran. Mol. Cell. Biol. 2006, 26, 1318–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, F.; Lin, X.; Chang, C.; Feng, X.H. Nuclear export of Smad2 and Smad3 by RanBP3 facilitates termination of TGF-β signaling. Dev. Cell 2009, 16, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Kang, Y.; Col, S.; Massagué, J. Smad2 nucleocytoplasmic shuttling by nucleoporins CAN/Nup214 and Nup153 feeds TGFβ signaling complexes in the cytoplasm and nucleus. Mol. Cell 2002, 10, 271–282. [Google Scholar] [CrossRef]
- Xu, L.; Alarcon, C.; Col, S.; Massague, J. Distinct domain utilization by Smad3 and Smad4 for nucleoporin interaction and nuclear import. J. Biol. Chem. 2003, 278, 42569–42577. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Ding, W.; Mulder, K.M. Requirement for the dynein light chain km23-1 in a Smad2-dependent transforming growth factor-β signaling pathway. J. Biol. Chem. 2007, 282, 19122–19132. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.Y.; Ding, W.; Mulder, K.M. The TGFβ receptor-interacting protein km23-1/DYNLRB1 plays an adaptor role in TGFβ1 autoinduction via its association with Ras. J. Biol. Chem. 2012, 287, 26453–26463. [Google Scholar] [CrossRef] [Green Version]
- Funaba, M.; Zimmerman, C.M.; Mathews, L.S. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002, 277, 41361–41368. [Google Scholar] [CrossRef] [Green Version]
- Yakymovych, I.; ten Dijke, P.; Heldin, C.H.; Souchelnytskyi, S. Regulation of Smad signaling by protein kinase C. FASEB J. 2001, 15, 553–555. [Google Scholar] [CrossRef]
- Saura, M.; Zaragoza, C.; Herranz, B.; Griera, M.; Diez-Marques, L.; Rodriguez-Puyol, D.; Rodriguez-Puyol, M. Nitric oxide regulates transforming growth factor-β signaling in endothelial cells. Circ. Res. 2005, 97, 1115–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Waddell, D.S.; Wang, W.; Wang, Z.; Liberati, N.T.; Yong, S.; Liu, X.; Wang, X.F. Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-β signaling. Oncogene 2008, 27, 7235–7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Zhang, J.; Sun, Q.; Tuazon, P.T.; Wu, X.; Traugh, J.A.; Chen, Y.G. p21-Activated kinase 2 (PAK2) inhibits TGF-β signaling in Madin-Darby canine kidney (MDCK) epithelial cells by interfering with the receptor-Smad interaction. J. Biol. Chem. 2012, 287, 13705–13712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelen, B.A.; Cohen, O.S.; Raychowdhury, M.K.; Chadee, D.N.; Zhang, Y.; Kyriakis, J.M.; Alessandrini, A.A.; Lin, H.Y. Phosphorylation of threonine 276 in Smad4 is involved in transforming growth factor-β-induced nuclear accumulation. Am. J. Physiol. Cell Physiol. 2003, 285, C823–C830. [Google Scholar] [CrossRef]
- Morén, A.; Raja, E.; Heldin, C.H.; Moustakas, A. Negative regulation of TGFβ signaling by the kinase LKB1 and the scaffolding protein LIP1. J. Biol. Chem. 2011, 286, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, H.A.; Jung, H.; Ha, H. Murine protein serine/threonine kinase 38 stimulates TGF-β signaling in a kinase-dependent manner via direct phosphorylation of Smad proteins. J. Biol. Chem. 2010, 285, 30959–30970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Duan, X.; Liang, Y.Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A functions as a Smad phosphatase to terminate TGFβ signaling. Cell 2006, 125, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pan, L.; Qin, X.; Chen, H.; Xu, Y.; Chen, Y.; Tang, H. MTMR4 attenuates transforming growth factor β (TGFβ) signaling by dephosphorylating R-Smads in endosomes. J. Biol. Chem. 2010, 285, 8454–8462. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, G.; Knockaert, M.; Alarcon, C.; Montalvo, E.; Brivanlou, A.H.; Massague, J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-β pathways. J. Biol. Chem. 2006, 281, 40412–40419. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.H.; Willis, D.; Long, J.; Liu, F.; Lin, X.; Feng, X.H. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-β signaling. J. Biol. Chem. 2006, 281, 38365–38375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chang, C.; Gehling, D.J.; Hemmati-Brivanlou, A.; Derynck, R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2001, 98, 974–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.Y.; Yamashita, M.; Coussens, N.P.; Tang, Y.; Wang, X.; Li, C.; Deng, C.X.; Cheng, S.Y.; Zhang, Y.E. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011, 30, 4777–4789. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-β pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragon, E.; Goerner, N.; Zaromytidou, A.I.; Xi, Q.; Escobedo, A.; Massague, J.; Macias, M.J. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Xu, X.; Li, L.; Ning, H.; Rong, Y.; Shang, Y.; Wang, Y.; Fu, X.Y.; Chang, Z. CHIP controls the sensitivity of transforming growth factor-β signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 2005, 280, 20842–20850. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, M.; Imamura, T.; Chiba, T.; Ebisawa, T.; Kawabata, M.; Tanaka, K.; Miyazono, K. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol. Biol. Cell 2001, 12, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Ramirez, A.; Waddell, D.S.; Li, Z.; Liu, X.; Wang, X.F. Axin and GSK3- control Smad3 protein stability and modulate TGF-β signaling. Genes Dev. 2008, 22, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Yang, C.; Hu, K.; Elly, C.; Liu, Y.C. Itch E3 ligase-mediated regulation of TGF-β signaling by modulating Smad2 phosphorylation. Mol. Cell 2004, 15, 825–831. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; Andrew, R.L.; Lee, K.L.; Petropoulou, C.; Dixon, J.E.; Navaratnam, N.; Norris, D.P.; Episkopou, V. Arkadia enhances Nodal/TGF-β signaling by coupling phospho-Smad2/3 activity and turnover. PLoS Biol. 2007, 5, e67. [Google Scholar] [CrossRef]
- Morén, A.; Hellman, U.; Inada, Y.; Imamura, T.; Heldin, C.H.; Moustakas, A. Differential ubiquitination defines the functional status of the tumor suppressor Smad4. J. Biol. Chem. 2003, 278, 33571–33582. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Liang, Y.Y.; Wrighton, K.; Ungermannova, D.; Wang, X.P.; Brunicardi, F.C.; Liu, X.; Feng, X.H.; Lin, X. Ubiquitination and proteolysis of cancer-derived Smad4 mutants by SCFSkp2. Mol. Cell. Biol. 2004, 24, 7524–7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agricola, E.; Randall, R.A.; Gaarenstroom, T.; Dupont, S.; Hill, C.S. Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 2011, 43, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Zacchigna, L.; Cordenonsi, M.; Soligo, S.; Adorno, M.; Rugge, M.; Piccolo, S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 2005, 121, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Manfrin, A.; Mamidi, A.; Martello, G.; Morsut, L.; Soligo, S.; Enzo, E.; Moro, S.; Polo, S.; Dupont, S.; et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell Biol. 2011, 13, 1368–1375. [Google Scholar] [CrossRef]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Melchior, F.; Feng, X.H.; Lin, X. Regulation of Smad4 sumoylation and transforming growth factor-β signaling by protein inhibitor of activated STAT1. J. Biol. Chem. 2004, 279, 22857–22865. [Google Scholar] [CrossRef] [Green Version]
- Imoto, S.; Sugiyama, K.; Muromoto, R.; Sato, N.; Yamamoto, T.; Matsuda, T. Regulation of transforming growth factor-β signaling by protein inhibitor of activated STAT, PIASy through Smad3. J. Biol. Chem. 2003, 278, 34253–34258. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Itoh, Y.; Abe, K.; Okamoto, T.; Daitoku, H.; Fukamizu, A.; Onozaki, K.; Hayashi, H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2007, 26, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Lönn, P.; van der Heide, L.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP-1 attenuates Smad-mediated transcription. Mol. Cell 2010, 40, 521–532. [Google Scholar] [CrossRef]
- Zawel, L.; Dai, J.L.; Buckhaults, P.; Zhou, S.; Kinzler, K.W.; Vogelstein, B.; Kern, S.E. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1998, 1, 611–617. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.H. Genome-wide mechanisms of Smad binding. Oncogene 2013, 32, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Martin-Malpartida, P.; Batet, M.; Kaczmarska, Z.; Freier, R.; Gomes, T.; Aragon, E.; Zou, Y.; Wang, Q.; Xi, Q.; Ruiz, L.; et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 2017, 8, 2070. [Google Scholar] [CrossRef] [PubMed]
- Aragon, E.; Wang, Q.; Zou, Y.; Morgani, S.M.; Ruiz, L.; Kaczmarska, Z.; Su, J.; Torner, C.; Tian, L.; Hu, J.; et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes Dev. 2019, 33, 1506–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Liu, X.; Ren, X.; Tian, Y.; Chen, Z.; Xu, X.; Du, Y.; Jiang, C.; Fang, Y.; Liu, Z.; et al. Smad2 and Smad3 have differential sensitivity in relaying TGFβ signaling and inversely regulate early lineage specification. Sci. Rep. 2016, 6, 21602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Loven, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.H.; Zhang, Y.; Wu, R.Y.; Derynck, R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes Dev. 1998, 12, 2153–2163. [Google Scholar] [CrossRef] [Green Version]
- Itoh, S.; Ericsson, J.; Nishikawa, J.-i.; Heldin, C.H.; ten Dijke, P. The transcriptional co-activator P/CAF potentiates TGF-β/Smad signaling. Nucl. Acid. Res. 2000, 28, 4291–4298. [Google Scholar] [CrossRef] [Green Version]
- Kahata, K.; Hayashi, M.; Asaka, M.; Hellman, U.; Kitagawa, H.; Yanagisawa, J.; Kato, S.; Imamura, T.; Miyazono, K. Regulation of transforming growth factor-β and bone morphogenetic protein signalling by transcriptional coactivator GCN5. Genes Cells 2004, 9, 143–151. [Google Scholar] [CrossRef]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Katsuno, Y.; Meyer, D.; Budi, E.H.; Chen, S.H.; Koeppen, H.; Wang, H.; Akhurst, R.J.; Derynck, R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018, 19, 135–155. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wu, H.; Xu, Y.; Zhang, Z.; Liu, T.; Lin, X.; Feng, X.H. Zinc finger protein 451 is a novel Smad corepressor in transforming growth factor-β signaling. J. Biol. Chem. 2014, 289, 2072–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Q.; Wang, Z.; Zaromytidou, A.I.; Zhang, X.H.; Chow-Tsang, L.F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A poised chromatin platform for TGF-β access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, V.; Sixt, K.M.; Gao, S.; Xu, X.; Huang, J.; Weigert, R.; Zhou, M.; Zhang, Y.E. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β. Mol. Cell 2016, 64, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 2010, 39, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [Green Version]
- Janakiraman, H.; House, R.P.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H.; Palanisamy, V. The Long (lncRNA) and Short (miRNA) of It: TGFβ-Mediated Control of RNA-Binding Proteins and Noncoding RNAs. Mol. Cancer Res. 2018, 16, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadee, C.; et al. The SMAD2/3 interactome reveals that TGFβ controls m(6)A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Mochizuki, T.; Miyazaki, H.; Hara, T.; Furuya, T.; Imamura, T.; Watabe, T.; Miyazono, K. Roles for the MH2 domain of Smad7 in the specific inhibition of transforming growth factor-β superfamily signaling. J. Biol. Chem. 2004, 279, 31568–31574. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-β signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Miyazono, K.; Miyazawa, K. Smad7 inhibits transforming growth factor-β family type I receptors through two distinct modes of interaction. J. Biol. Chem. 2010, 285, 30804–30813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [Green Version]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-β (TGF-β) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-β (transforming growth factor-β) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-β type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Itoh, S.; Landström, M.; Hermansson, A.; Itoh, F.; Heldin, C.H.; Heldin, N.E.; ten Dijke, P. Transforming growth factor β1 induces nuclear export of inhibitory Smad7. J. Biol. Chem. 1998, 273, 29195–29201. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhang, J.; Pan, L.; Wang, P.; Xue, H.; Zhang, L.; Gao, X.; Zhao, X.; Ning, Y.; Chen, Y.G. TSC-22 promotes transforming growth factor β-mediated cardiac myofibroblast differentiation by antagonizing Smad7 activity. Mol. Cell. Biol. 2011, 31, 3700–3709. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.H.; Meng, A.; Chen, Y.G. Smad7 antagonizes transforming growth factor β signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell. Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Cao, X. A nuclear antagonistic mechanism of inhibitory Smads in transforming growth factor-β signaling. J. Biol. Chem. 2002, 277, 4176–4182. [Google Scholar] [CrossRef] [Green Version]
- Pulaski, L.; Landström, M.; Heldin, C.H.; Souchelnytskyi, S. Phosphorylation of Smad7 at Ser-249 does not interfere with its inhibitory role in transforming growth factor-β-dependent signaling but affects Smad7-dependent transcriptional activation. J. Biol. Chem. 2001, 276, 14344–14349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grönroos, E.; Hellman, U.; Heldin, C.H.; Ericsson, J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell 2002, 10, 483–493. [Google Scholar] [CrossRef]
- Kume, S.; Haneda, M.; Kanasaki, K.; Sugimoto, T.; Araki, S.; Isshiki, K.; Isono, M.; Uzu, T.; Guarente, L.; Kashiwagi, A.; et al. SIRT1 inhibits transforming growth factor β-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J. Biol. Chem. 2007, 282, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsson, M.; Heldin, C.H.; Ericsson, J.; Grönroos, E. The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef] [Green Version]
- Elkouris, M.; Kontaki, H.; Stavropoulos, A.; Antonoglou, A.; Nikolaou, K.C.; Samiotaki, M.; Szantai, E.; Saviolaki, D.; Brown, P.J.; Sideras, P.; et al. SET9-Mediated Regulation of TGF-β Signaling Links Protein Methylation to Pulmonary Fibrosis. Cell Rep. 2016, 15, 2733–2744. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Winawer, S.; Friedman, E. Two different signal transduction pathways can be activated by transforming growth factor β1 in epithelial cells. J. Biol. Chem. 1994, 269, 13231–13237. [Google Scholar]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Chen, Y.G. Specific activation of mitogen-activated protein kinase by transforming growth factor-β receptors in lipid rafts is required for epithelial cell plasticity. Mol. Biol. Cell 2009, 20, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Minden, A.; Martinetto, H.; Claret, F.X.; Lange-Carter, C.; Mercurio, F.; Johnson, G.L.; Karin, M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science 1995, 268, 286–290. [Google Scholar] [CrossRef]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent SMAD and JNK signaling in transforming growth factor-β-mediated transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, K.; Shirakabe, K.; Shibuya, H.; Irie, K.; Oishi, I.; Ueno, N.; Taniguchi, T.; Nishida, E.; Matsumoto, K. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 1995, 270, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landström, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.I.; Kwak, J.H.; Na, H.J.; Kim, J.K.; Ding, Y.; Choi, M.E. Transforming growth factor-b (TGF-b1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-β receptor kinase activity in mesangial cells. J. Biol. Chem. 2009, 284, 22285–22296. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landström, M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, F.F.; de Vinuesa, A.G.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.H.; Bauer, A.; Rousseau, A.; et al. TRAF4 Promotes TGF-β Receptor Signaling and Drives Breast Cancer Metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, A.; von Bulow, V.; Hamidi, R.; Winssinger, N.; Barluenga, S.; Heldin, C.H.; Landström, M. Polyubiquitination of transforming growth factor β (TGFβ)-associated kinase 1 mediates nuclear factor-κB activation in response to different inflammatory stimuli. J. Biol. Chem. 2012, 287, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Yumoto, K.; Thomas, P.S.; Lane, J.; Matsuzaki, K.; Inagaki, M.; Ninomiya-Tsuji, J.; Scott, G.J.; Ray, M.K.; Ishii, M.; Maxson, R.; et al. TGF-β-activated Kinase 1 (Tak1) Mediates Agonist-induced Smad Activation and Linker Region Phosphorylation in Embryonic Craniofacial Neural Crest-derived Cells. J. Biol. Chem. 2013, 288, 13467–13480. [Google Scholar] [CrossRef] [Green Version]
- Edlund, S.; Bu, S.; Schuster, N.; Aspenström, P.; Heuchel, R.; Heldin, N.E.; ten Dijke, P.; Heldin, C.H.; Landström, M. Transforming growth factor-β1 (TGF-β)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-β-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 2003, 14, 529–544. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.M.; Lee, J.H.; Park, J.; Oh, Y.S.; Lee, S.K.; Park, J.S.; Lee, Y.S.; Kim, J.H.; Lee, J.Y.; Bae, Y.S.; et al. Smad6 inhibits non-canonical TGF-β1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat. Commun. 2013, 4, 2562. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I transforming growth factor β receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Fang, R.; Wang, X.F.; Zhang, F.; Chen, D.Y.; Zhou, B.; Wang, H.S.; Cai, S.H.; Du, J. Stabilization of Snail through AKT/GSK-3β signaling pathway is required for TNF-α-induced epithelial-mesenchymal transition in prostate cancer PC3 cells. Eur. J. Pharmacol. 2013, 714, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Godoy, P.; Bachmann, A.; Liu, Y.; Barzan, D.; Ilkavets, I.; Maier, P.; Herskind, C.; Hengstler, J.G.; Dooley, S. Distinct role of endocytosis for Smad and non-Smad TGF-β signaling regulation in hepatocytes. J. Hepatol. 2011, 55, 369–378. [Google Scholar] [CrossRef]
- Liu, C.; Xu, P.; Lamouille, S.; Xu, J.; Derynck, R. TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell 2009, 35, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFβ type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [Green Version]
- Sundar, R.; Gudey, S.K.; Heldin, C.H.; Landström, M. TRAF6 promotes TGFβ-induced invasion and cell-cycle regulation via Lys63-linked polyubiquitination of Lys178 in TGFβ type I receptor. Cell Cycle 2015, 14, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.H.; Landström, M. TRAF6 stimulates the tumor-promoting effects of TGFβ type I receptor through polyubiquitination and activation of presenilin 1. Sci. Signal. 2014, 7, ra2. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Tomcik, M.; Palumbo-Zerr, K.; Distler, A.; Beyer, C.; Lang, V.; Horn, A.; Zerr, P.; Zwerina, J.; Gelse, K.; et al. JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor β in systemic sclerosis. Arthritis Rheum. 2012, 64, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-β (TGF-β) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef]
- Edlund, S.; Landström, M.; Heldin, C.H.; Aspenström, P. Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Li, J.; Hu, P.P.; Waddell, D.; Zhang, J.; Wang, X.F. The activity of guanine exchange factor NET1 is essential for transforming growth factor-β-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 15362–15368. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, E.; Vasilaki, E.; Vorvis, C.; Iliopoulos, D.; Moustakas, A.; Kardassis, D.; Stournaras, C. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-β and miR-24: Role in epithelial-to-mesenchymal transition. Oncogene 2012, 31, 2862–2875. [Google Scholar] [CrossRef]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef]
- Wilkes, M.C.; Murphy, S.J.; Garamszegi, N.; Leof, E.B. Cell-type-specific activation of PAK2 by transforming growth factor β independent of Smad2 and Smad3. Mol. Cell. Biol. 2003, 23, 8878–8889. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.W.; Zhu, Y.Q.; Sharma, K. Transforming growth factor-β1 stimulates protein kinase A in mesangial cells. J. Biol. Chem. 1998, 273, 8522–8527. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Duan, C.J.; Binkley, C.; Li, G.; Uhler, M.D.; Logsdon, C.D.; Simeone, D.M. A transforming growth factor β-induced Smad3/Smad4 complex directly activates protein kinase A. Mol. Cell. Biol. 2004, 24, 2169–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, R.C.; Knittle, A.M.; Nagaraj, N.; van Dinther, M.; Choudhary, C.; ten Dijke, P.; Mann, M.; Sharma, K. Time-resolved dissection of early phosphoproteome and ensuing proteome changes in response to TGF-β. Sci. Signal. 2014, 7, rs5. [Google Scholar] [PubMed] [Green Version]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, A.K.; Petrovic, N.; Moodley, Y.P.; Fogel-Petrovic, M.; Kroeger, K.M.; Seeber, R.M.; Eidne, K.A.; Thompson, P.J.; Knight, D.A. αvβ3 Integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. J. Biol. Chem. 2004, 279, 37726–37733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M., Jr.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-β induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef]
- Remy, I.; Montmarquette, A.; Michnick, S.W. PKB/Akt modulates TGF-β signalling through a direct interaction with Smad3. Nat. Cell Biol. 2004, 6, 358–365. [Google Scholar] [CrossRef]
- Seong, H.A.; Jung, H.; Choi, H.S.; Kim, K.T.; Ha, H. Regulation of transforming growth factor-β signaling and PDK1 kinase activity by physical interaction between PDK1 and serine-threonine kinase receptor-associated protein. J. Biol. Chem. 2005, 280, 42897–42908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Kim, B.C.; Tognon, C.; Lee, H.J.; Patel, S.; Lannon, C.L.; Maris, J.M.; Triche, T.J.; Sorensen, P.H.; Kim, S.J. The ETV6-NTRK3 chimeric tyrosine kinase suppresses TGF-β signaling by inactivating the TGF-β type II receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 16239–16244. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Yun, C.; Kwak, M.K.; Kim, T.A.; Kim, S.J. TrkC binds to the type II TGF-β receptor to suppress TGF-β signaling. Oncogene 2007, 26, 7684–7691. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Szabolcs, A.; Dutta, S.K.; Yaqoob, U.; Jagavelu, K.; Wang, L.; Leof, E.B.; Urrutia, R.A.; Shah, V.H.; Mukhopadhyay, D. Neuropilin-1 Mediates Divergent R-Smad Signaling and the Myofibroblast Phenotype. J. Biol. Chem. 2010, 285, 31840–31848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Hiemer, S.E.; Szymaniak, A.D.; Varelas, X. The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells. J. Biol. Chem. 2014, 289, 13461–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-β-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Park, J.O.; Kim, T.S.; Kim, S.K.; Kim, T.H.; Kim, M.C.; Park, G.S.; Kim, J.H.; Kuninaka, S.; Olson, E.N.; et al. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat. Commun. 2016, 7, 11961. [Google Scholar] [CrossRef]
- Nallet-Staub, F.; Yin, X.; Gilbert, C.; Marsaud, V.; Ben Mimoun, S.; Javelaud, D.; Leof, E.B.; Mauviel, A. Cell density sensing alters TGF-β signaling in a cell-type-specific manner, independent from Hippo pathway activation. Dev. Cell 2015, 32, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Felici, A.; Wurthner, J.U.; Parks, W.T.; Giam, L.R.; Reiss, M.; Karpova, T.S.; McNally, J.G.; Roberts, A.B. TLP, a novel modulator of TGF-β signaling, has opposite effects on Smad2- and Smad3-dependent signaling. EMBO J. 2003, 22, 4465–4477. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. https://doi.org/10.3390/biom10030487
Tzavlaki K, Moustakas A. TGF-β Signaling. Biomolecules. 2020; 10(3):487. https://doi.org/10.3390/biom10030487
Chicago/Turabian StyleTzavlaki, Kalliopi, and Aristidis Moustakas. 2020. "TGF-β Signaling" Biomolecules 10, no. 3: 487. https://doi.org/10.3390/biom10030487
APA StyleTzavlaki, K., & Moustakas, A. (2020). TGF-β Signaling. Biomolecules, 10(3), 487. https://doi.org/10.3390/biom10030487