2-Oxoester Phospholipase A2 Inhibitors with Enhanced Metabolic Stability




Abstract
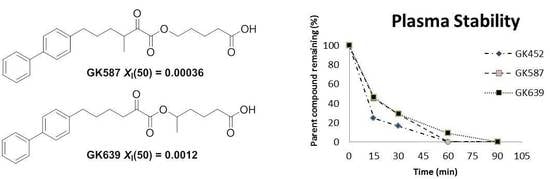
:
1. Introduction
2. Materials and Methods
2.1. General Chemistry Methods
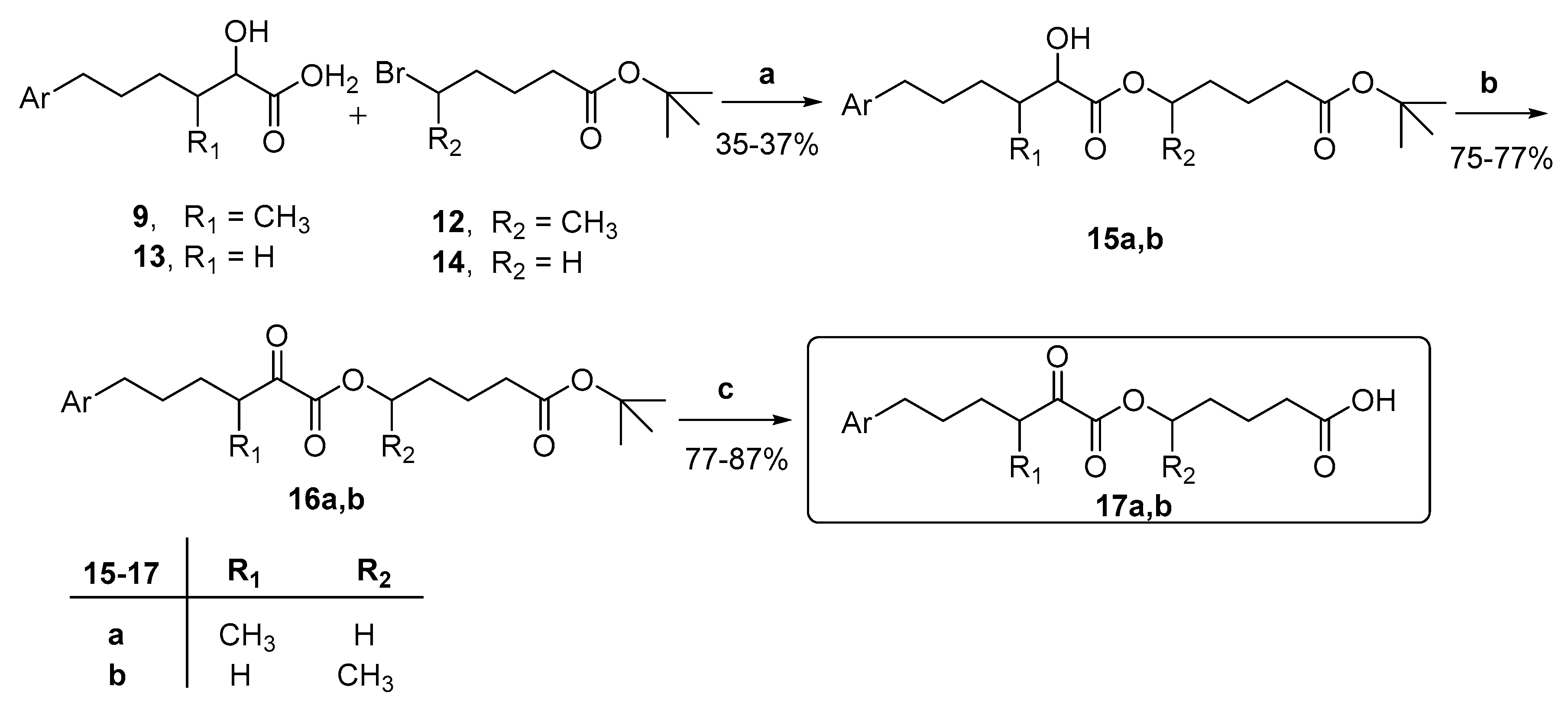
- General procedure for the synthesis of 2-hydroxy esters 15a,b.
- General procedure for the synthesis of 2-oxoesters 16a,b.
- General procedure for the synthesis of 2-oxoesters 17a,b.
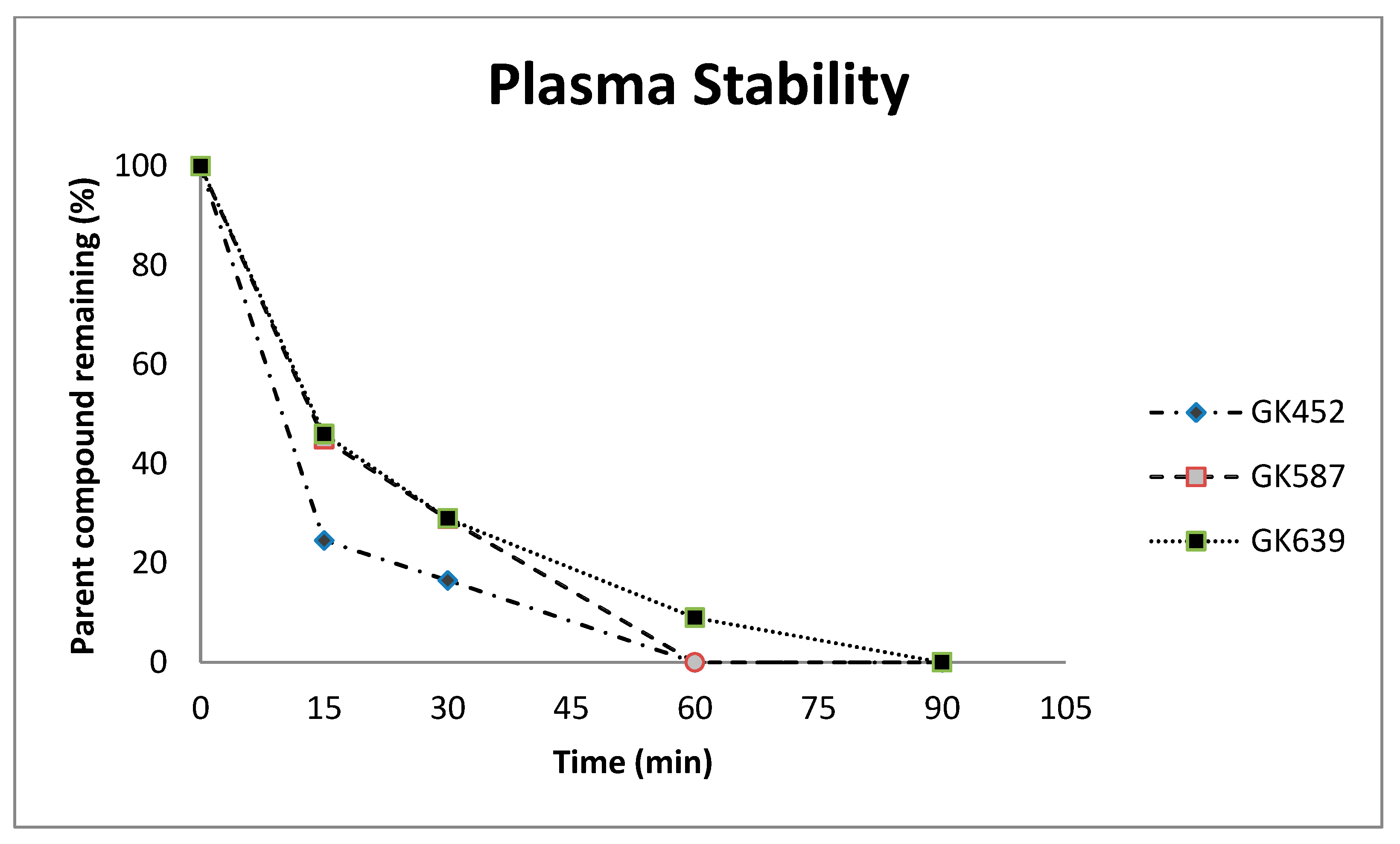
2.2. Plasma Stability Studies
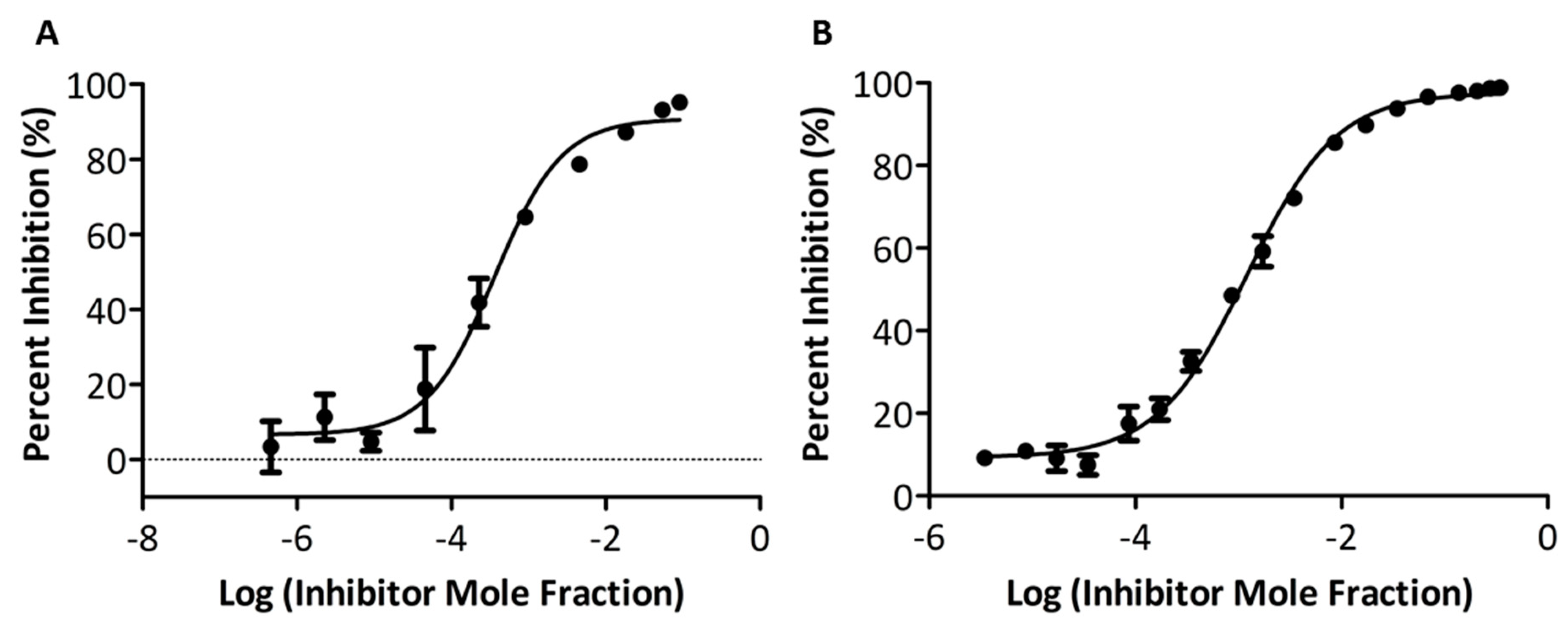
2.3. In vitro PLA2 Activity Assay
3. Results and Discussion
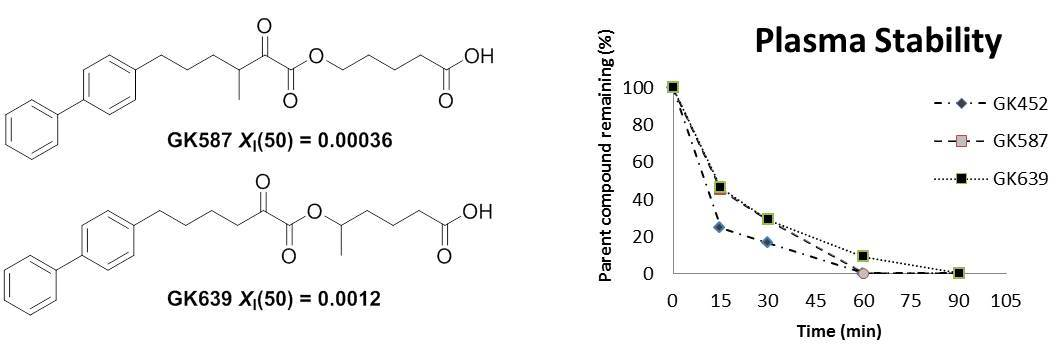
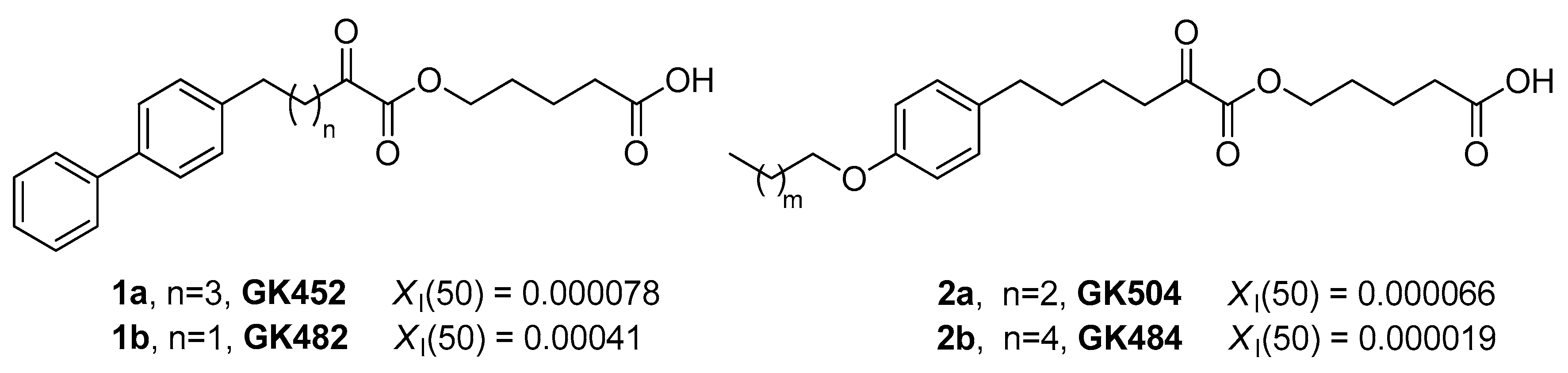
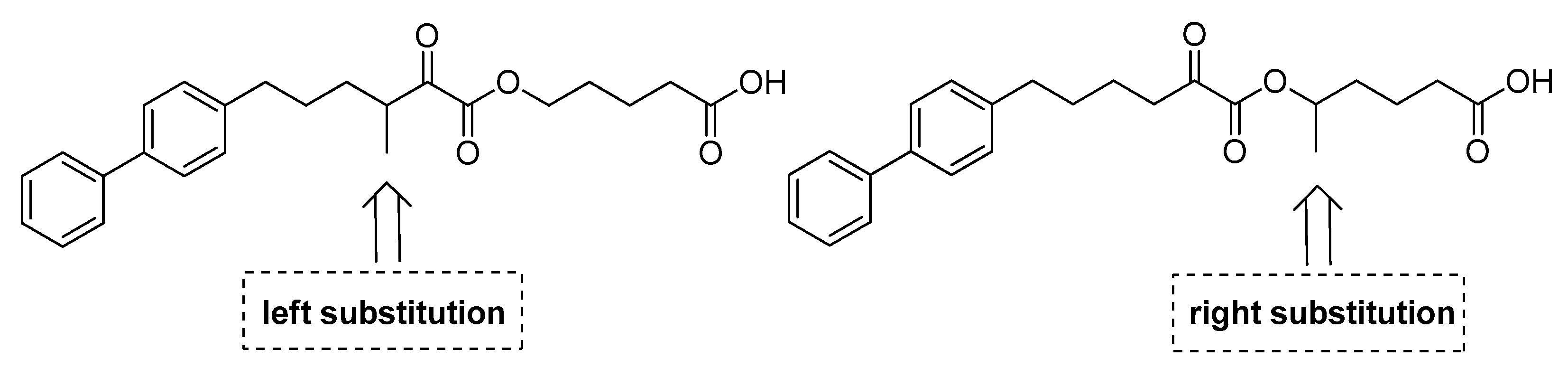
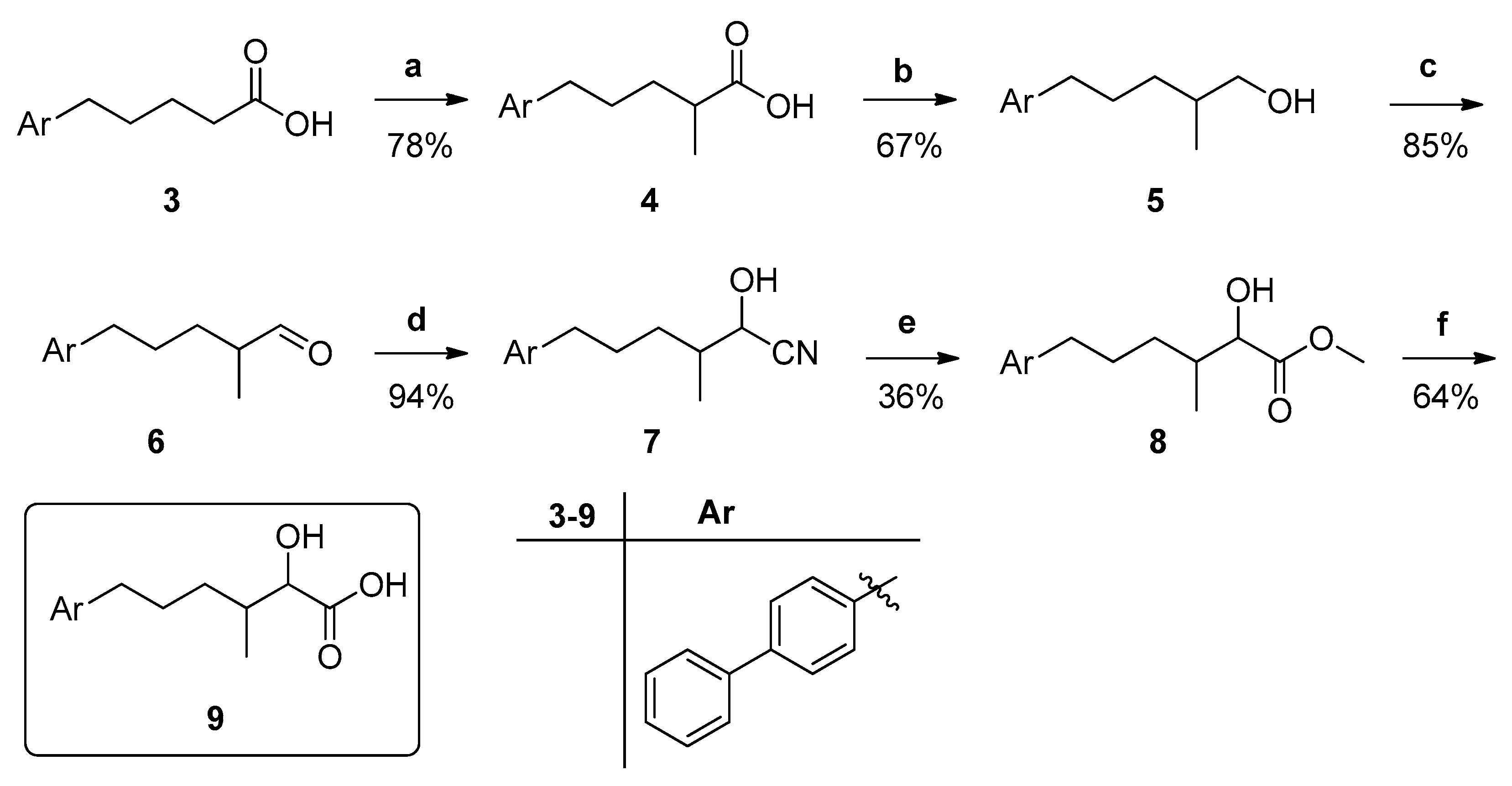
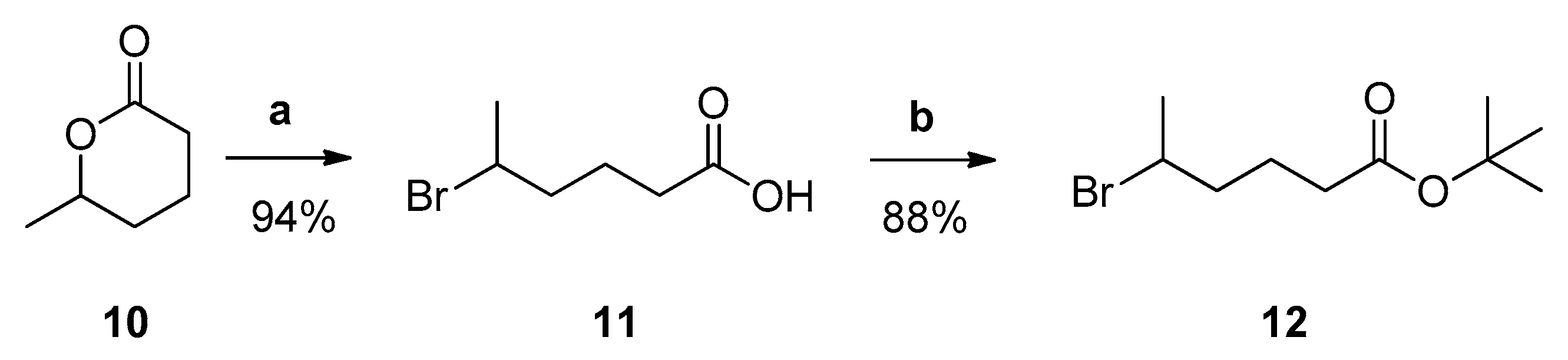
3.1. Design and Synthesis of Inhibitors
3.2. Plasma Stability Studies
3.3. In Vitro Inhibitory Potency and Selectivity of Compounds GK587 and GK639
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Nakatani, Y.; Atsumi, G.; Inoue, K.; Kudo, I. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 2017, 37, 121–179. [Google Scholar] [CrossRef]
- Vasquez, A.M.; Mouchlis, D.V.; Dennis, E.A. Review of four major distinct types of human phospholipase A2. Adv. Biol. Regul. 2018, 67, 212–218. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Chen, Y.; McCammon, J.A.; Dennis, E.A. Membrane allostery and unique hydrophobic sites promote enzyme substrate specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, Y.; Shindou, H.; Shimizu, T. Cytosolic phospholipase A2 and lysophospholipid acyltransferases. BBA Mol. Cell Biol. Lipids 2019, 1864, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Huang, Z.; Taheri, M.R.; O’Leary, E.; Li, E.; Moskowitz, M.A.; Sapirstein, A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 1997, 390, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Uozumi, N.; Kume, K.; Nagase, T.; Nakatani, N.; Ishii, S.; Tashiro, F.; Komagata, Y.; Maki, K.; Ikuta, K.; Ouchi, Y.; et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 1997, 390, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Chuang, D.Y.; Zong, Y.; Jiang, J.; Lee, J.C.M.; Gu, Z.; Simonyi, A. Role of cytosolic phospholipase A2 in oxidative and inflammatory signaling pathways in different cell types in the central nervous system. Mol. Neurobiol. 2014, 50, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Li, R.; Greenlief, C.M.; Fritsche, K.L.; Gu, Z.; Cui, J.; Lee, J.C.; Beversdorf, D.Q.; Sun, G.Y. Unveiling anti-oxidative and anti-inflammatory effects of docosahexaenoic acid and its lipid peroxidation product on lipopolysaccharide-stimulated BV-2 microglial cells. J. Neuroinfl. 2018, 15, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Fritsche, K.L.; Beversdorf, D.Q.; Gu, Z.; Lee, J.C.; Folk, W.R.; Greenlief, C.M.; Sun, G.Y. Yin-Yang mechanisms regulating lipid peroxidation of docosahexaenoic acid and arachidonic acid in the central nervous system. Front. Neurol. 2019, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Kokotou, M.G.; Limnios, D.; Nikolaou, A.; Psarra, A.; Kokotos, G. Inhibitors of phospholipase A2 and their therapeutic potential: An update on patents (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. BBA Mol. Cell Biol. Lipids 2019, 1864, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, G.; Six, D.A.; Loukas, V.; Smith, T.; Constantinou-Kokotou, V.; Hadjipavlou-Litina, D.; Kotsovolou, S.; Chiou, A.; Beltzner, C.C.; Dennis, E.A. Inhibition of group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells and in vivo. J. Med. Chem. 2004, 47, 3615–3628. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.; Barbayanni, E.; Constantinou-Kokotou, V.; Peristeraki, A.; Six, D.A.; Cooper, J.; Harkewicz, R.; Deems, R.A.; Dennis, E.A.; Kokotos, G. Differential inhibition of group IVA and group VIA phospholipases A2 by 2-oxoamides. J. Med. Chem. 2006, 49, 2821–2828. [Google Scholar] [CrossRef] [Green Version]
- Six, D.A.; Barbayanni, E.; Loukas, V.; Constantinou-Kokotou, V.; Hadjipavlou-Litina, D.; Stephens, D.; Wong, A.C.; Magrioti, V.; Moutevelis-Minakakis, P.; Baker, S.F.; et al. Structure-activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phospholipase A2. J. Med. Chem. 2007, 50, 4222–4235. [Google Scholar] [CrossRef]
- Baskakis, C.; Magrioti, V.; Cotton, N.; Stephens, D.; Constantinou-Kokotou, V.; Dennis, E.A.; Kokotos, G. Synthesis of polyfluoro ketones for selective inhibition of human phospholipase A2 enzymes. J. Med. Chem. 2008, 51, 8027–8037. [Google Scholar] [CrossRef] [Green Version]
- Kokotos, G.; Hsu, Y.-H.; Burke, J.E.; Baskakis, C.; Kokotos, C.G.; Magrioti, V.; Dennis, E.A. Potent and selective fluoroketone inhibitors of group VIA calcium-independent phospholipase A2. J. Med. Chem. 2010, 53, 3602–3610. [Google Scholar] [CrossRef] [Green Version]
- Kokotos, G.; Feuerherm, A.J.; Barbayianni, E.; Shah, I.; Sæther, M.; Magrioti, V.; Nguyen, T.; Constantinou-Kokotou, V.; Dennis, E.A.; Johansen, B. Inhibition of group IVA cytosolic phospholipase A2 by thiazolyl ketones in vitro, ex vivo, and in vivo. J. Med. Chem. 2014, 57, 7523–7535. [Google Scholar] [CrossRef]
- Kalyvas, A.; Baskakis, C.; Magrioti, V.; Constantinou-Kokotou, V.; Stephens, D.; Lopez-Vales, R.; Lu, J.Q.; Yong, V.W.; Dennis, E.A.; Kokotos, G.; et al. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain 2009, 132, 1221–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bone, R.N.; Gai, Y.; Magrioti, V.; Kokotou, M.G.; Ali, T.; Lei, X.; Tse, H.M.; Kokotos, G.; Ramanadham, S. Inhibition of Ca2+-independent phospholipase A2β (iPLA2β) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 2015, 64, 541–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokotou, M.G.; Galiatsatou, G.; Magrioti, V.; Koutoulogenis, G.; Barbayianni, E.; Limnios, D.; Mouchlis, V.D.; Satpathy, B.; Navratil, A.; Dennis, E.A.; et al. 2-Oxoesters: A novel class of potent and selective inhibitors of cytosolic group IVA phospholipase A2. Sci. Rep. 2017, 7, 7025. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.; Kokotou, M.G.; Galiatsatou, G.; Mouchlis, V.D.; Dennis, E.A.; Kokotos, G. Highly potent 2-oxoester inhibitors of cytosolic phospholipase A2 (GIVA cPLA2). ACS Omega 2018, 3, 8843–8853. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Armando, A.M.; Dennis, E.A. Substrate specific inhibition constants for phospholipase A2 acting on unique phospholipid substrates in mixed micelles and membranes using lipidomics. J. Med. Chem. 2019, 62, 1999–2007. [Google Scholar] [CrossRef]
- Sun, S.; Fu, J. Methyl-containing pharmaceuticals: Methylation in drug design. Bioorg. Med. Chem. Lett. 2018, 28, 3283–3289. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Wan, S.-L.; Chen, D.; Liu, Q.R.; Ding, C.-H.; Fang, P.; Hou, X.-L. Enantioselective construction of quaternary carbon stereocenter via palladium-catalyzed asymmetric allylic alkylation of lactones. Synthesis 2016, 48, 1568–1572. [Google Scholar] [CrossRef]
- Kokotos, G. A convenient one-pot conversion of N-protected amino acids and peptides into alcohols. Synthesis 1990, 1990, 299–301. [Google Scholar] [CrossRef]
- Haruki, S.; Akemi, N.; Mamoru, K. A new one-pot synthetic method for selenium containing medium-sized α,β-unsaturated cyclic ketones. Synthesis 2008, 2008, 3229–3236. [Google Scholar]
- Foster, D.A. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochim. Biophys. Acta 2009, 1791, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | GIVA cPLA2 | GIVA iPLA2 | GIVA sPLA2 | |
|---|---|---|---|---|---|
| % Inhibitiona | XI(50) | % Inhibitiona | % Inhibitiona | ||
| GK452 |  | >95% | 0.000078 ± 0.00001b | N.D.b,c | N.D.b,c |
| GK587 |  | >95% | 0.00036 ± 0.00007 | N.D.c | N.D.c |
| GK639 |  | >95% | 0.0012 ± 0.00008 | N.D.c | N.D.c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koutoulogenis, G.S.; Kokotou, M.G.; Hayashi, D.; Mouchlis, V.D.; Dennis, E.A.; Kokotos, G. 2-Oxoester Phospholipase A2 Inhibitors with Enhanced Metabolic Stability. Biomolecules 2020, 10, 491. https://doi.org/10.3390/biom10030491
Koutoulogenis GS, Kokotou MG, Hayashi D, Mouchlis VD, Dennis EA, Kokotos G. 2-Oxoester Phospholipase A2 Inhibitors with Enhanced Metabolic Stability. Biomolecules. 2020; 10(3):491. https://doi.org/10.3390/biom10030491
Chicago/Turabian StyleKoutoulogenis, Giorgos S., Maroula G. Kokotou, Daiki Hayashi, Varnavas D. Mouchlis, Edward A. Dennis, and George Kokotos. 2020. "2-Oxoester Phospholipase A2 Inhibitors with Enhanced Metabolic Stability" Biomolecules 10, no. 3: 491. https://doi.org/10.3390/biom10030491
APA StyleKoutoulogenis, G. S., Kokotou, M. G., Hayashi, D., Mouchlis, V. D., Dennis, E. A., & Kokotos, G. (2020). 2-Oxoester Phospholipase A2 Inhibitors with Enhanced Metabolic Stability. Biomolecules, 10(3), 491. https://doi.org/10.3390/biom10030491