Structural Dynamics of the Lipid Antigen-Binding Site of CD1d Protein


 and
and
Abstract
:1. Introduction
2. Materials and Methods
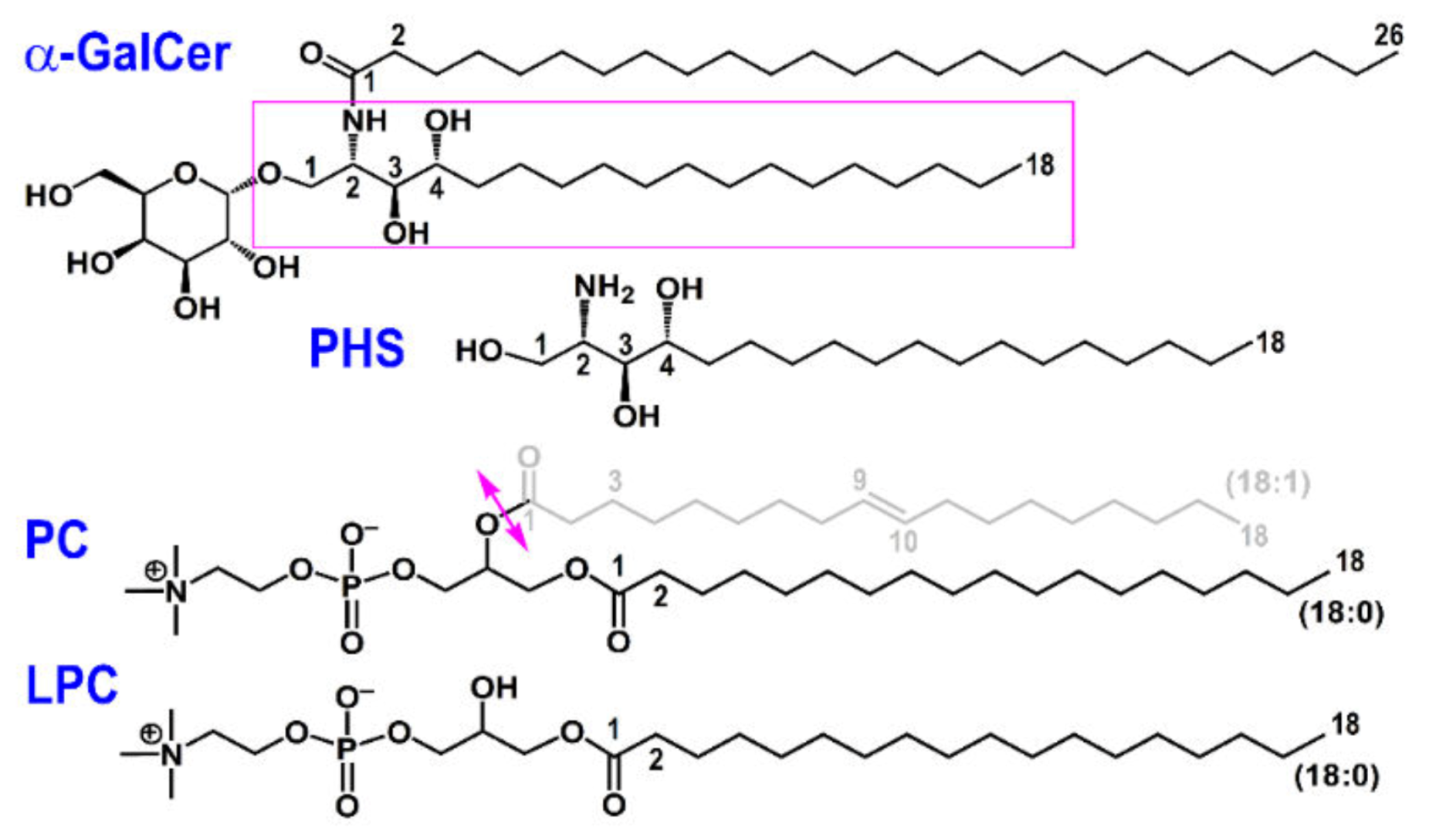
2.1. Initial Geometries of Lipid Antigens and Proteins
2.2. Docking Calculations to Set Initial Geometries of Protein–Ligand and Protein–Protein Complexes
2.3. Protonation States and Poisson-Boltzmann Electrostatic Potentials
2.4. Molecular Dynamics (MD) Calculations
2.5. Calculation of Molecular Properties
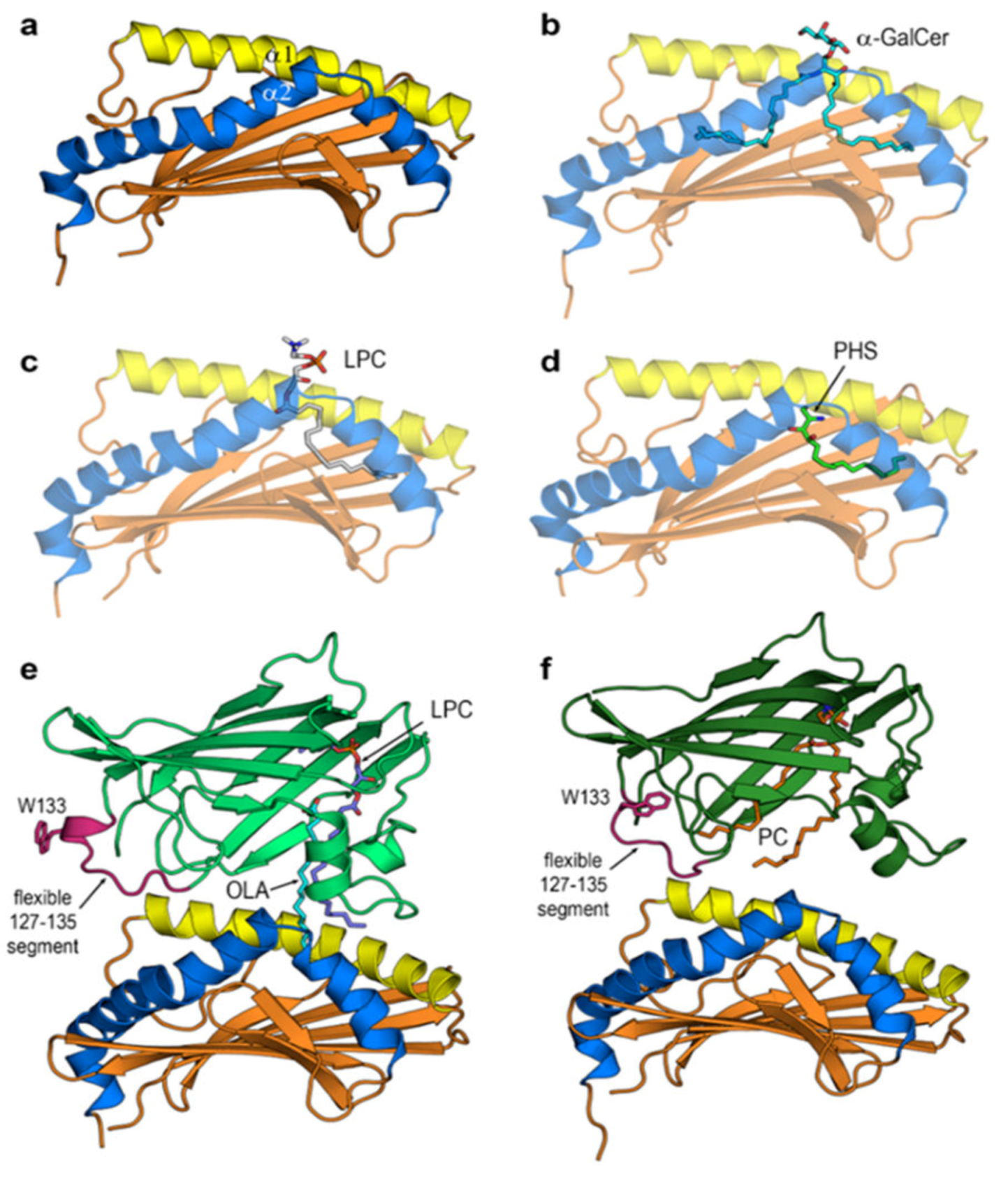
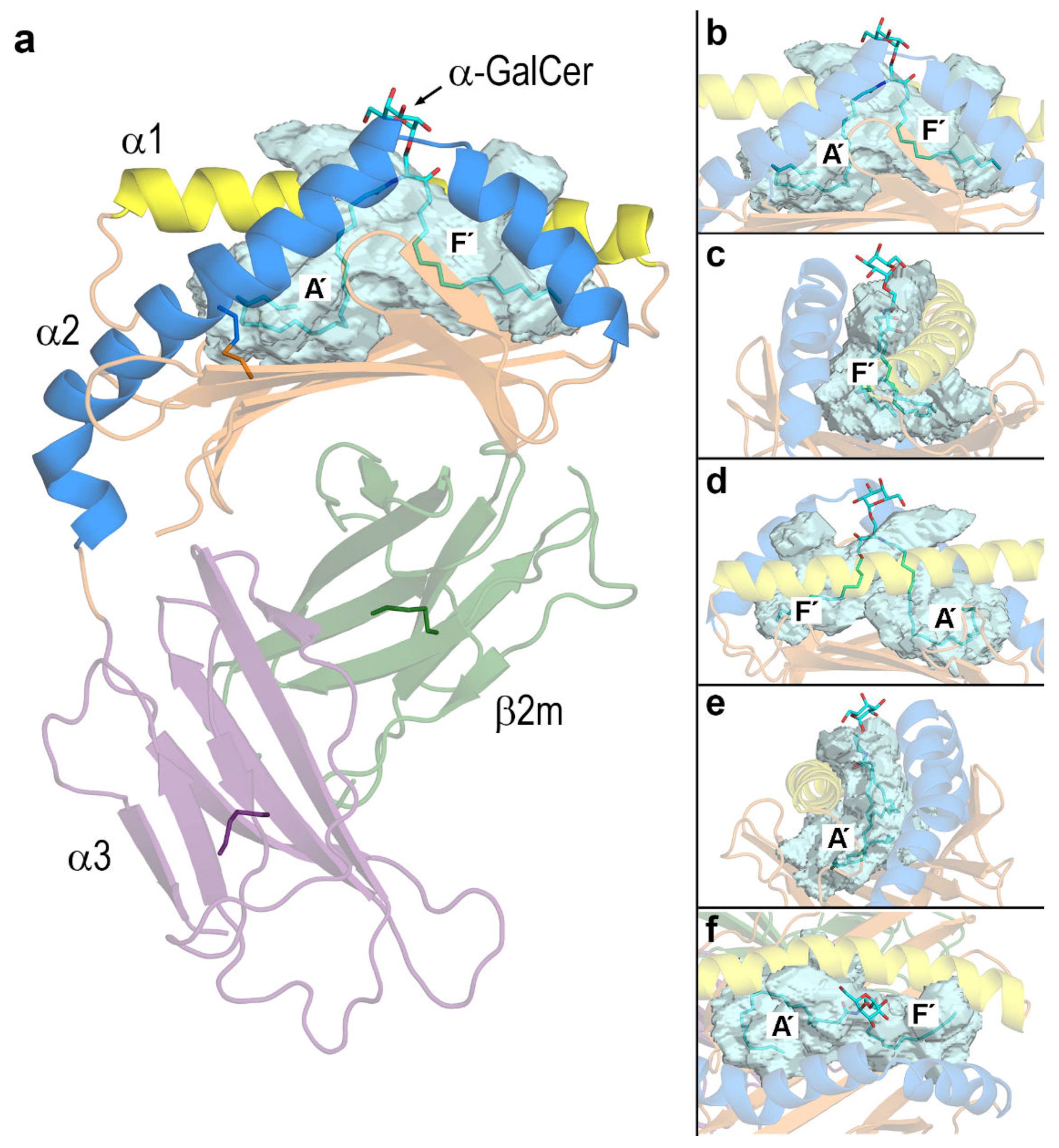
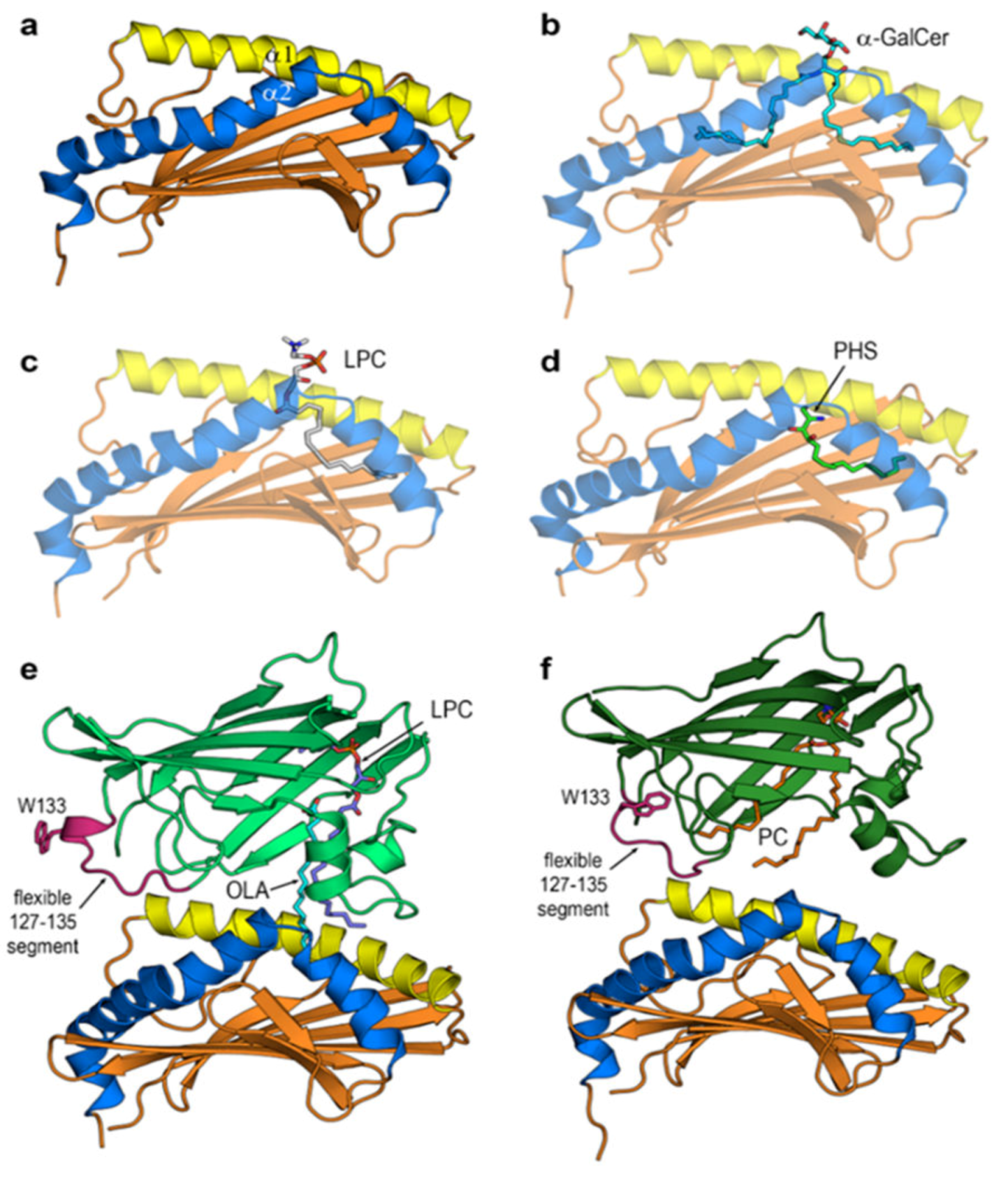
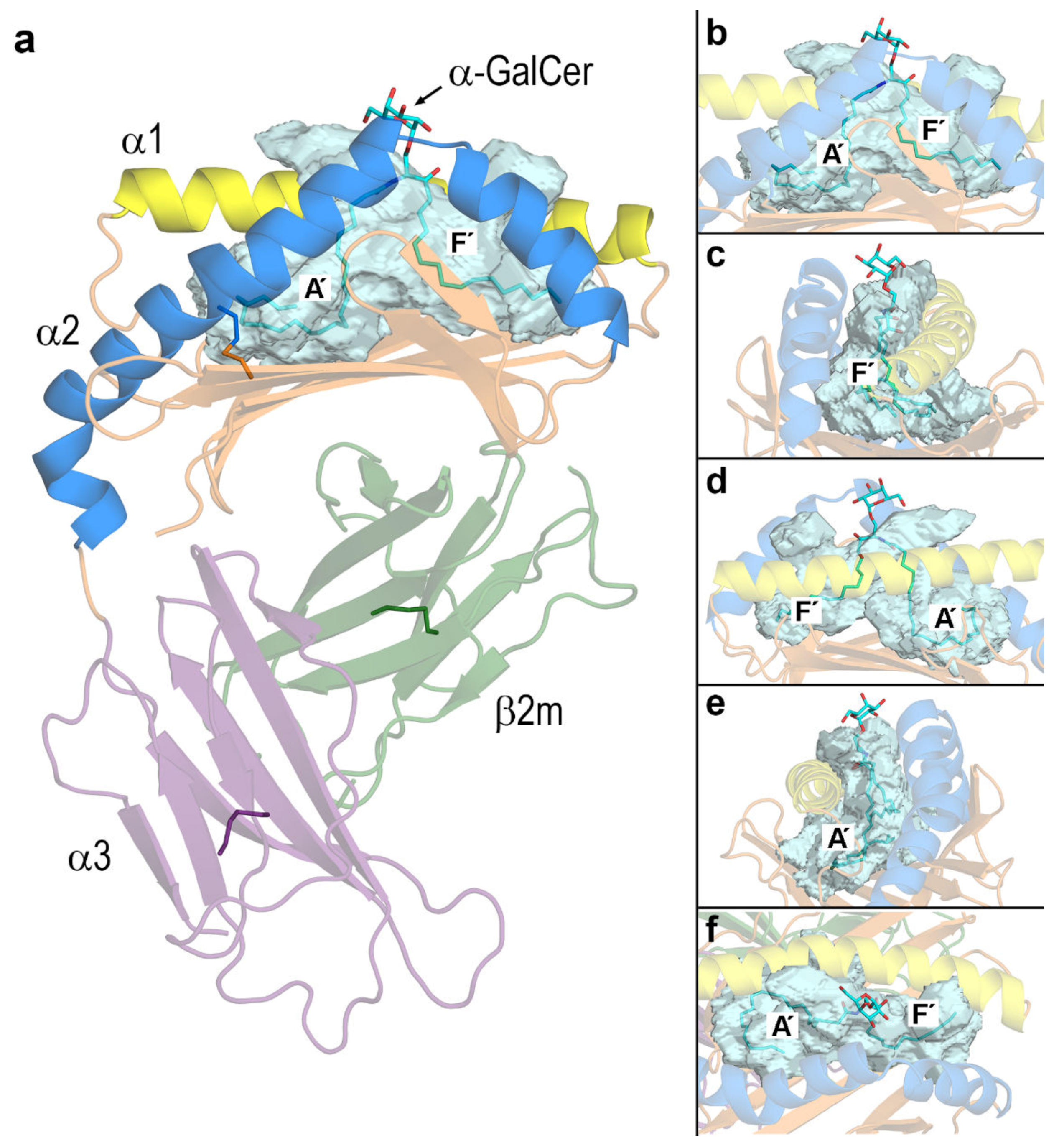
3. Results
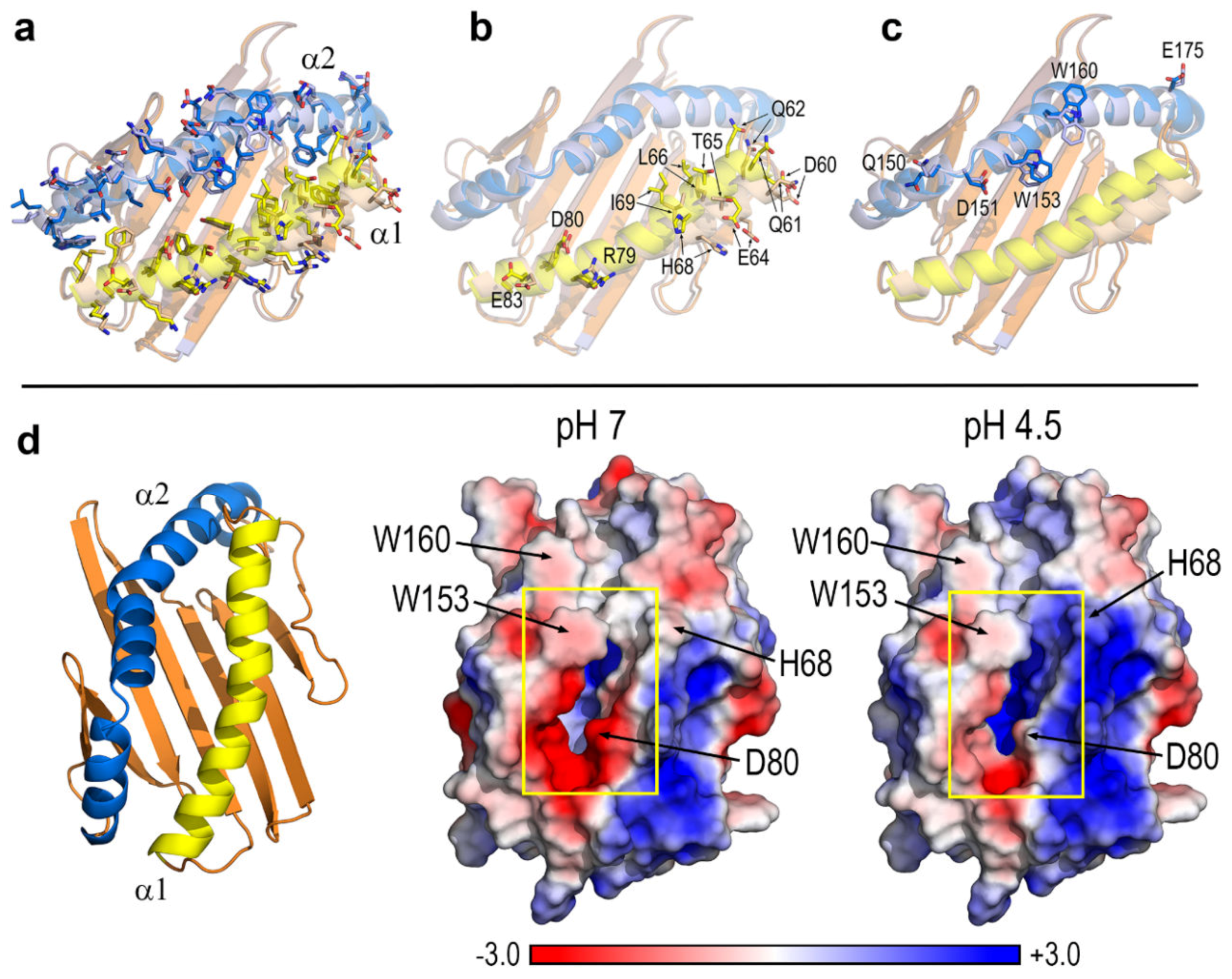
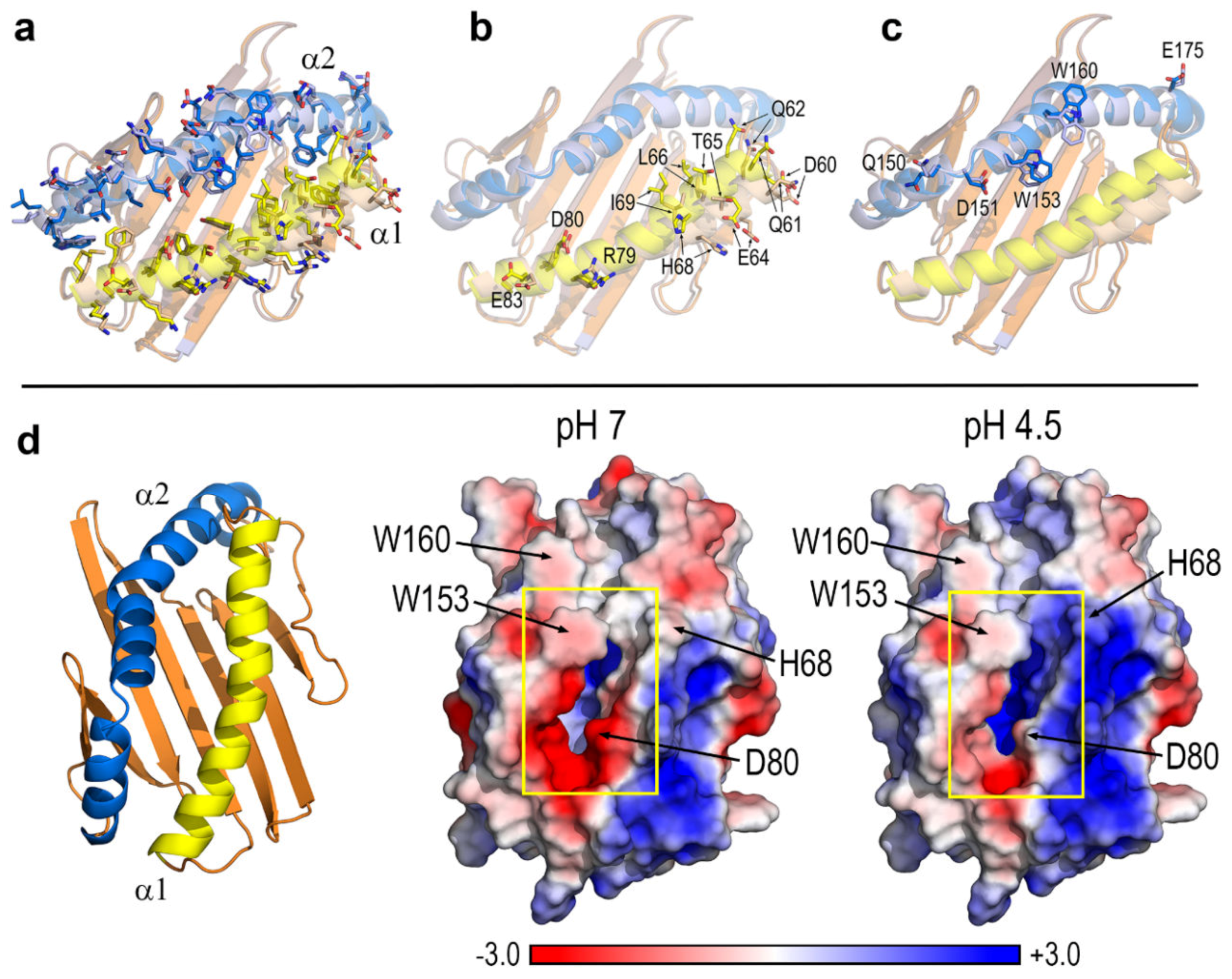
3.1. Key Residues and pH-Dependent Electrostatics in the Portal Entrance to Lipid–Antigen Channels of CD1d
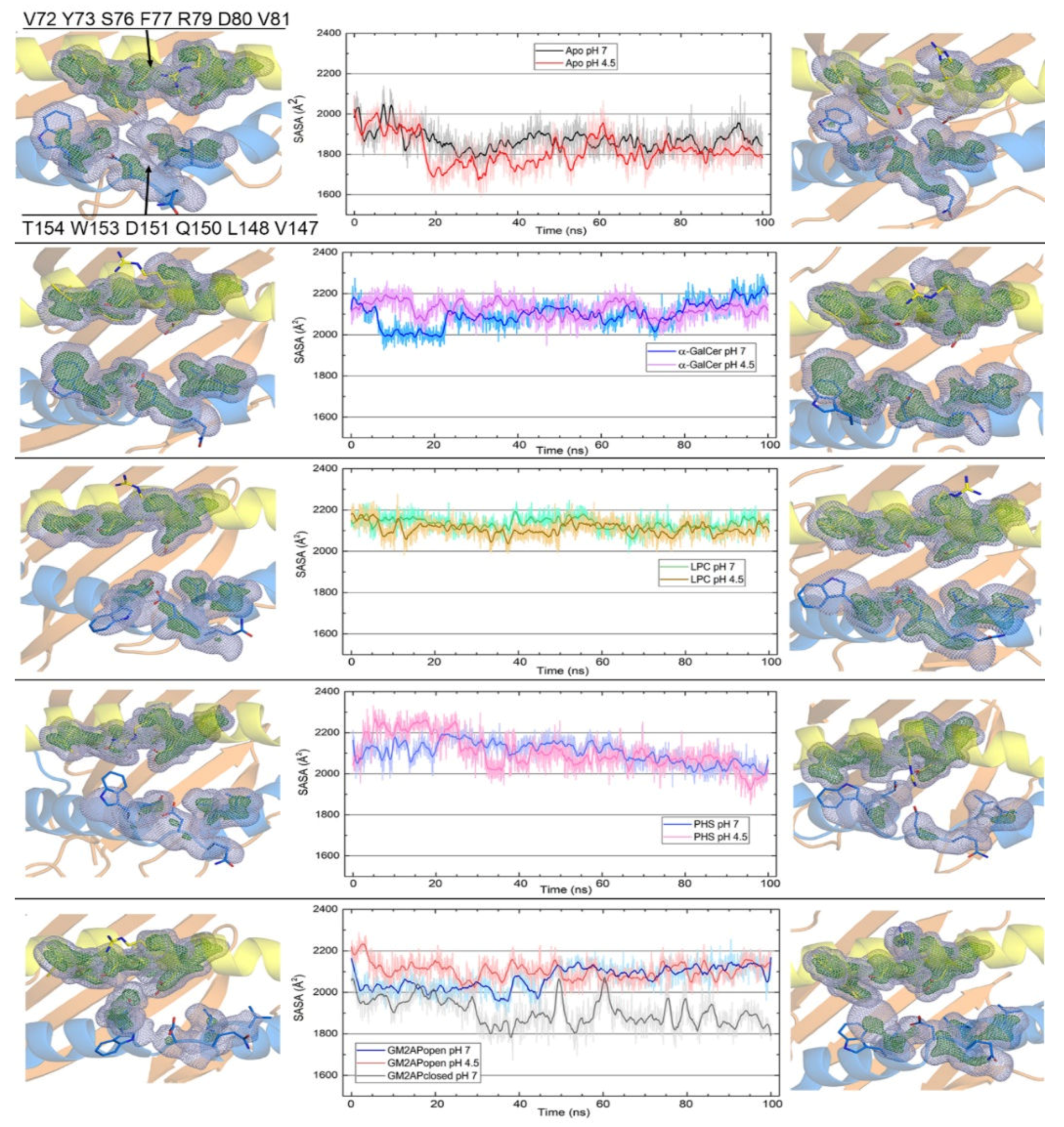
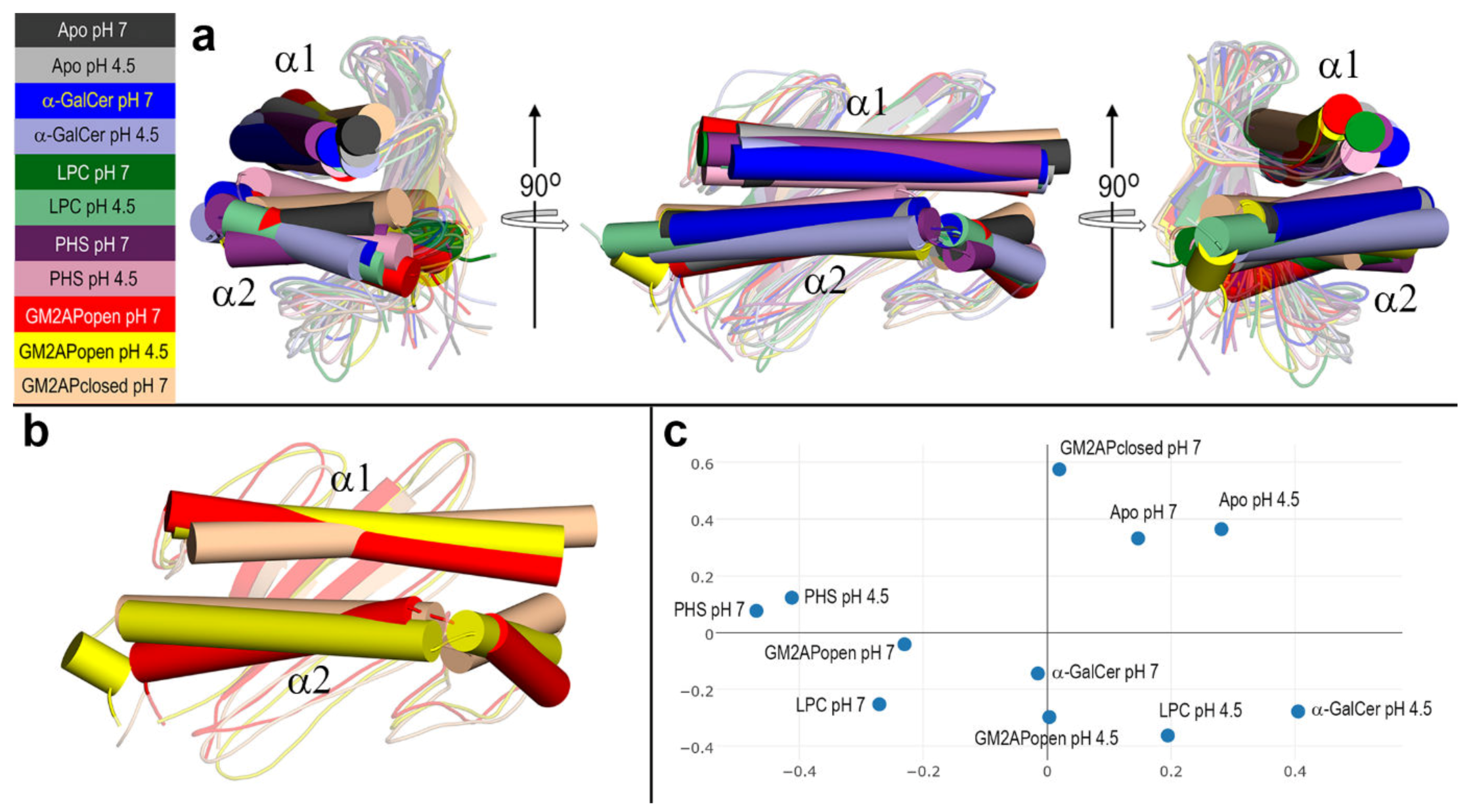
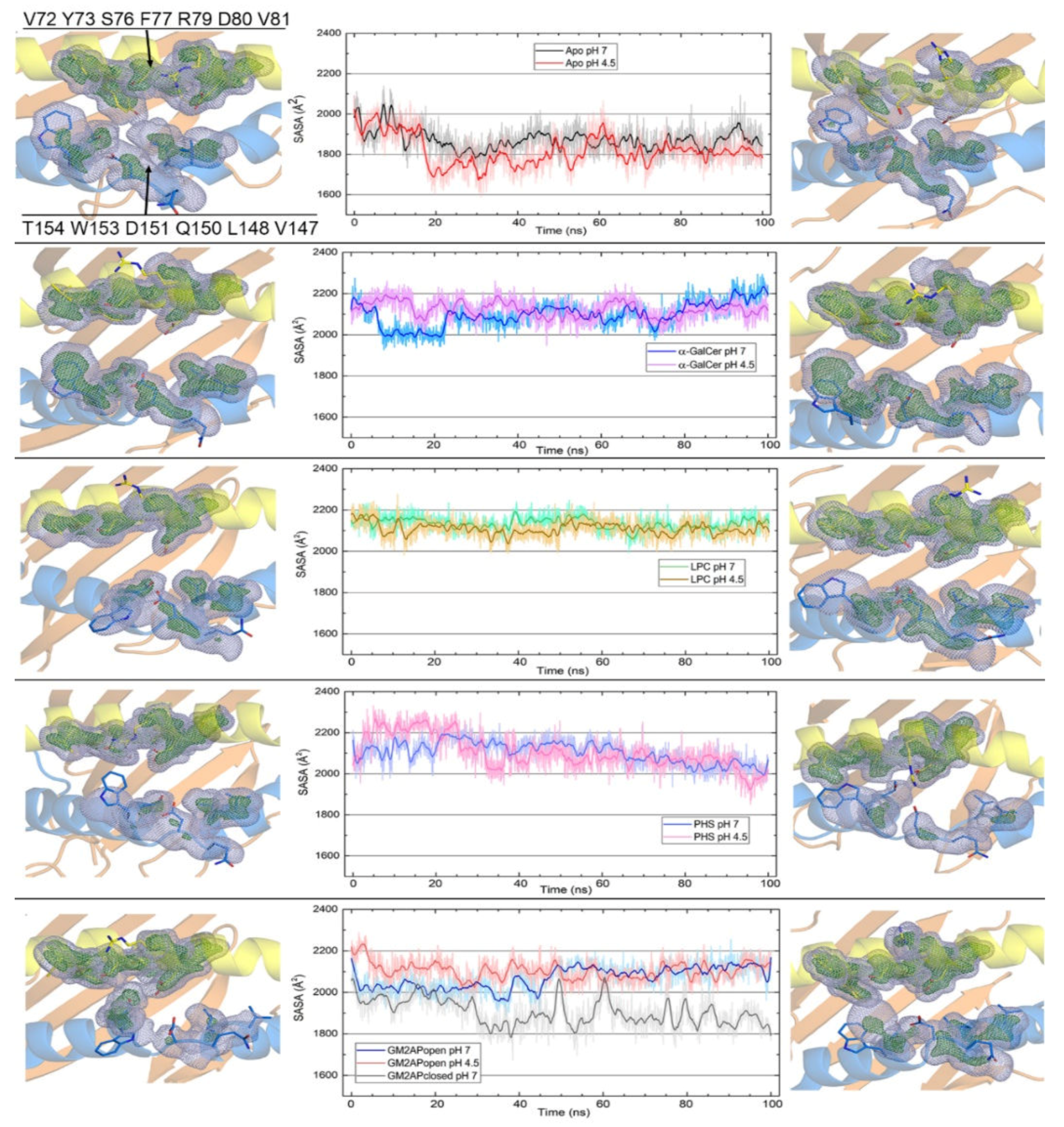
3.2. Dynamic Changes in the Portal Entrance
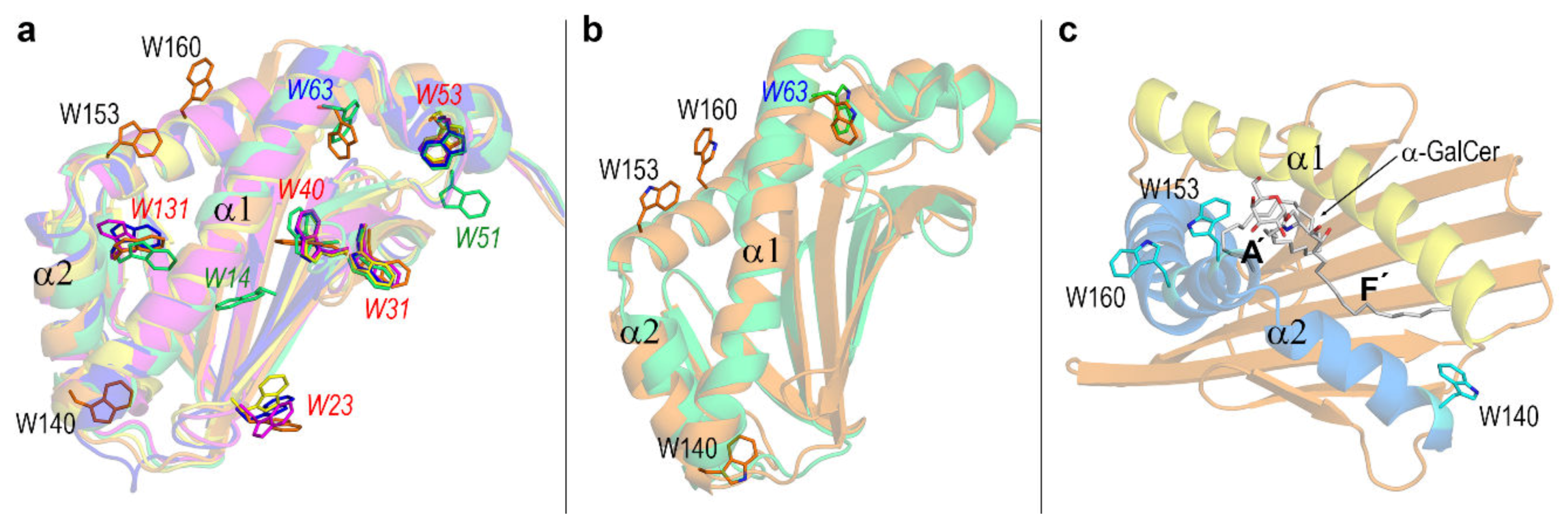
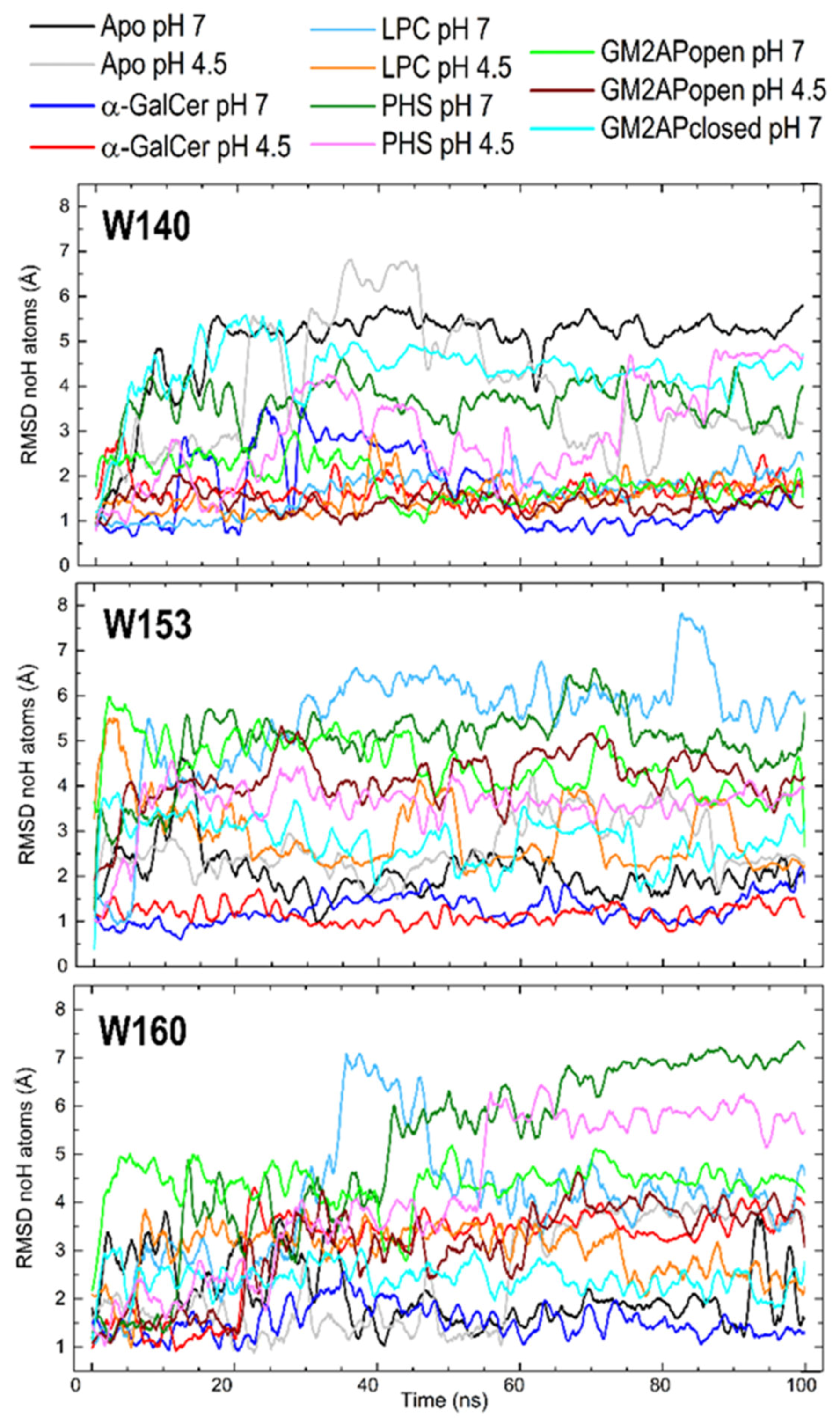
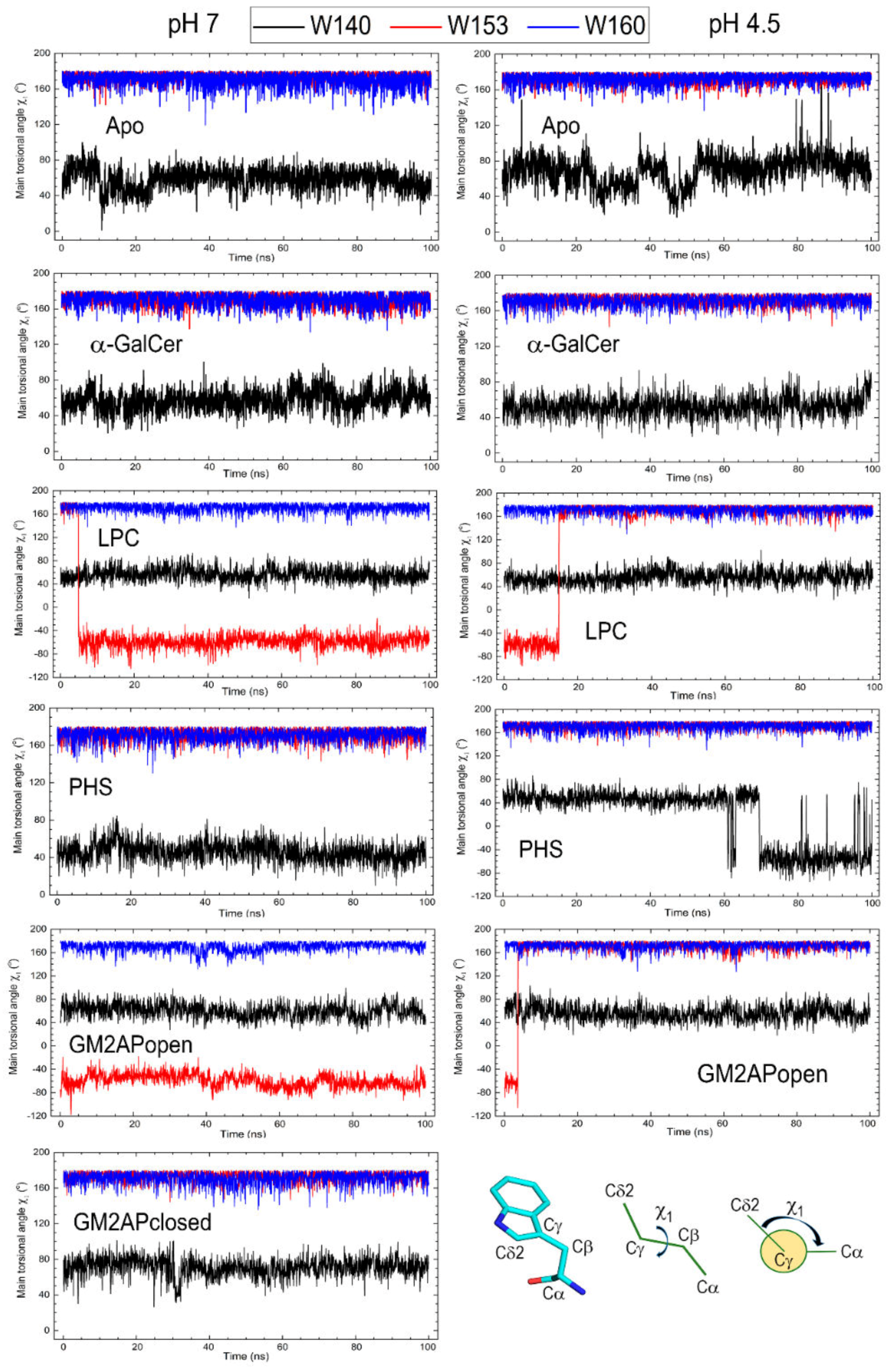
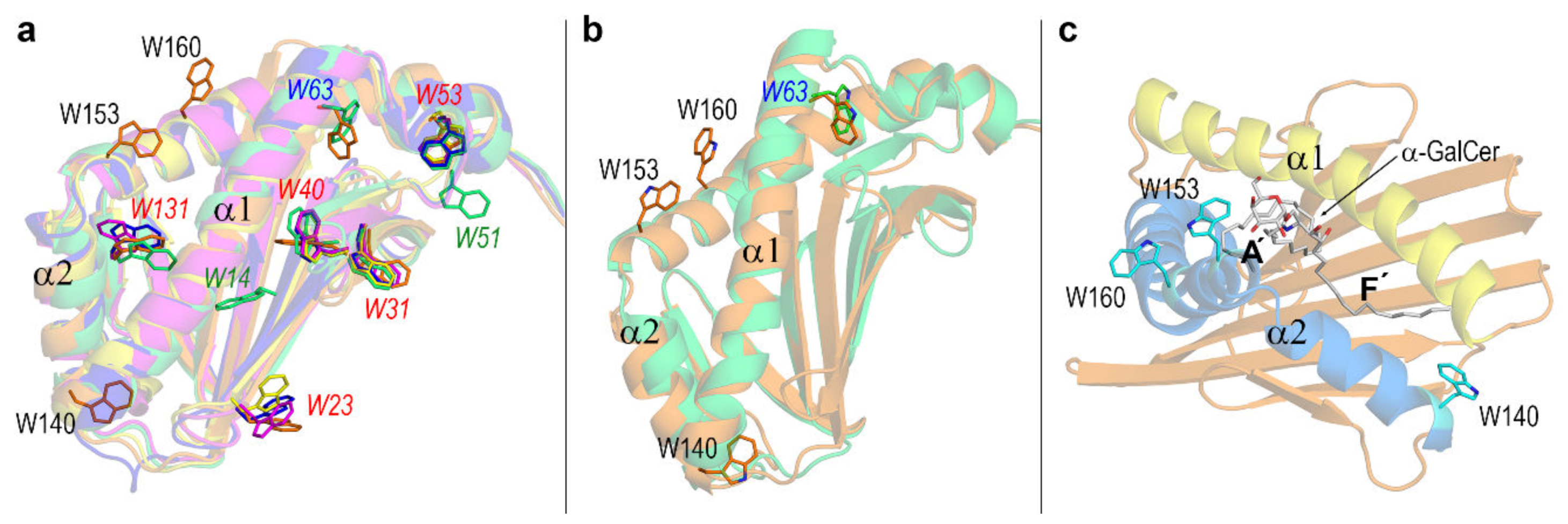
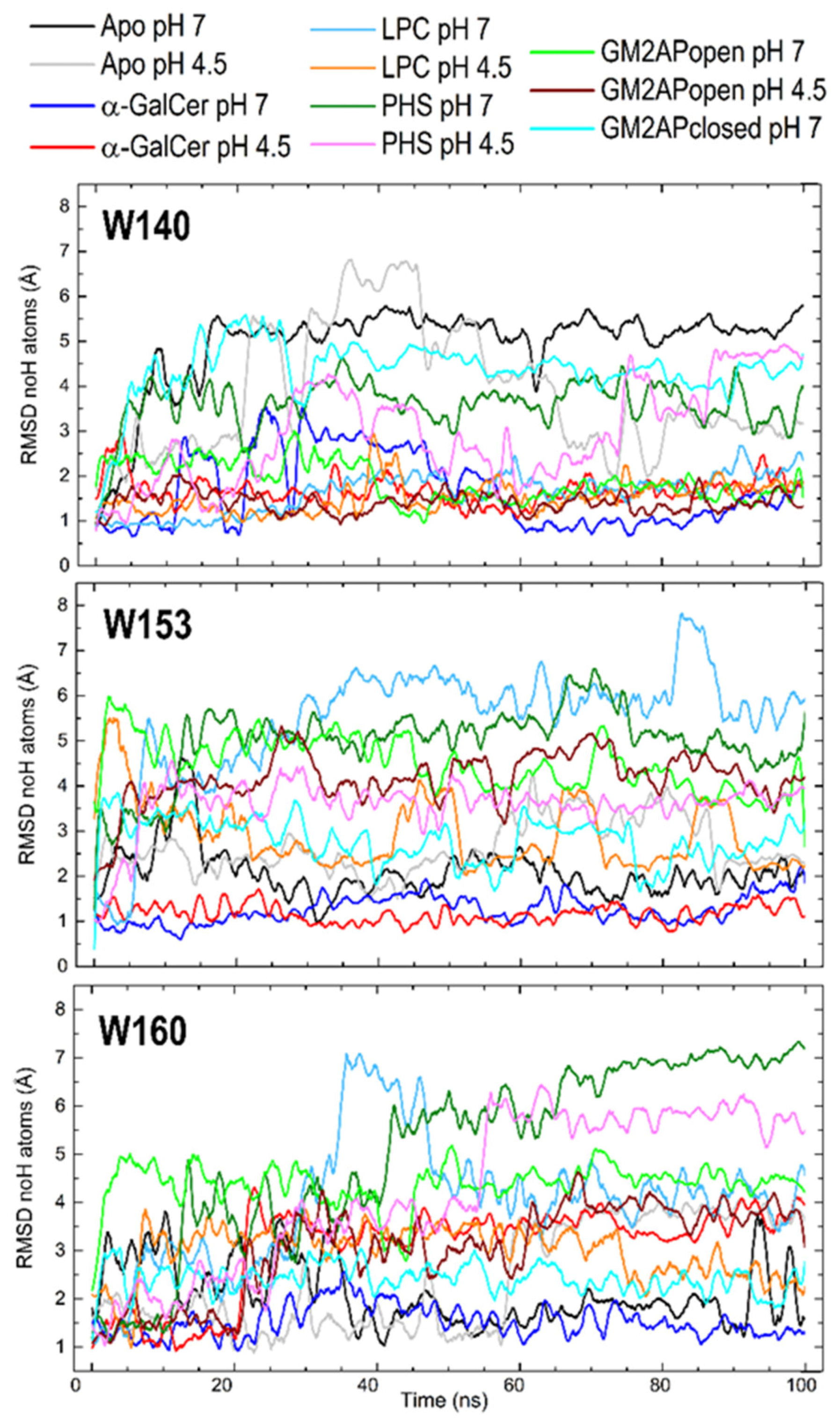
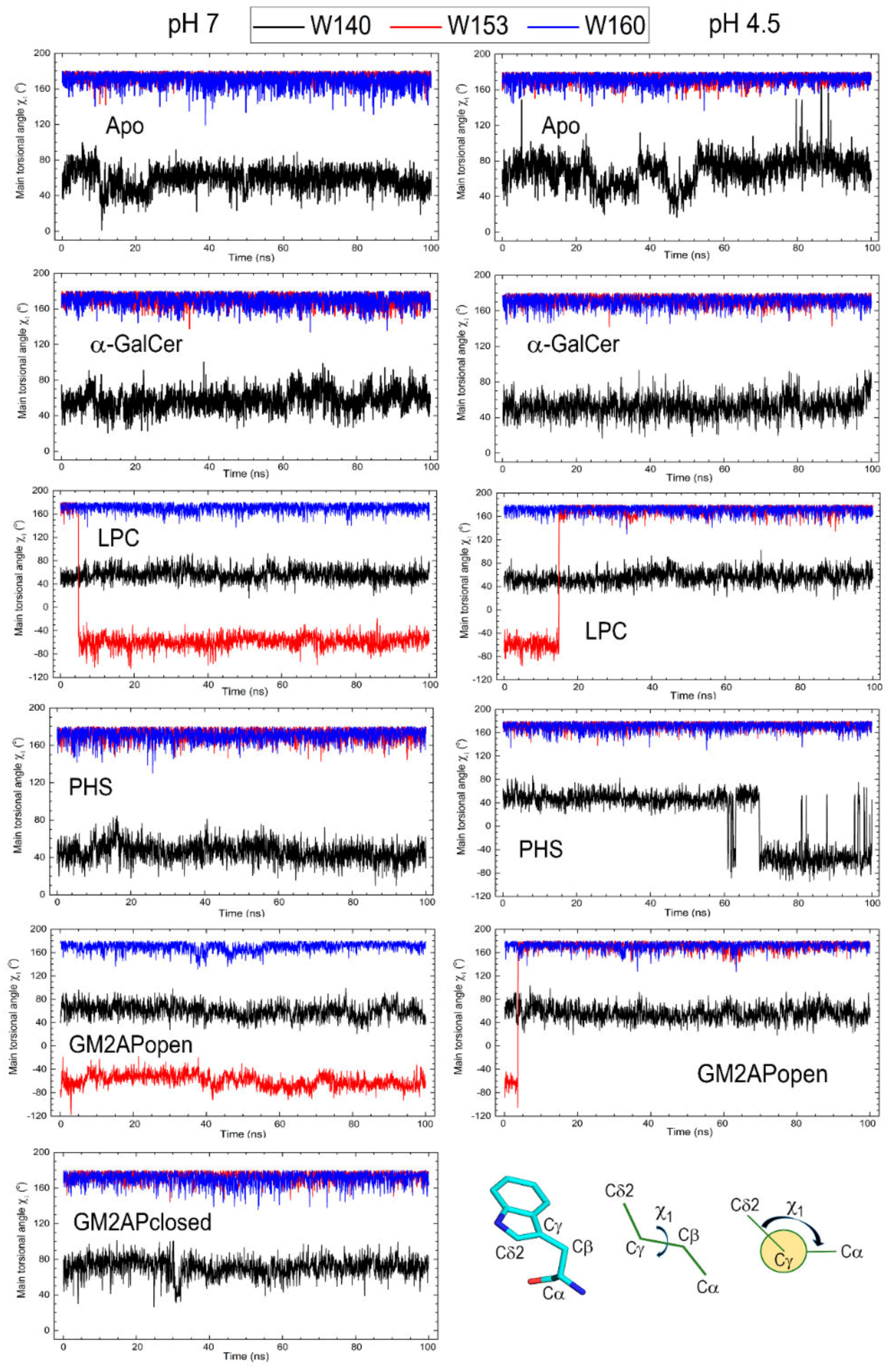
3.3. Dynamic Changes in Three Exposed Tryptophans of CD1d
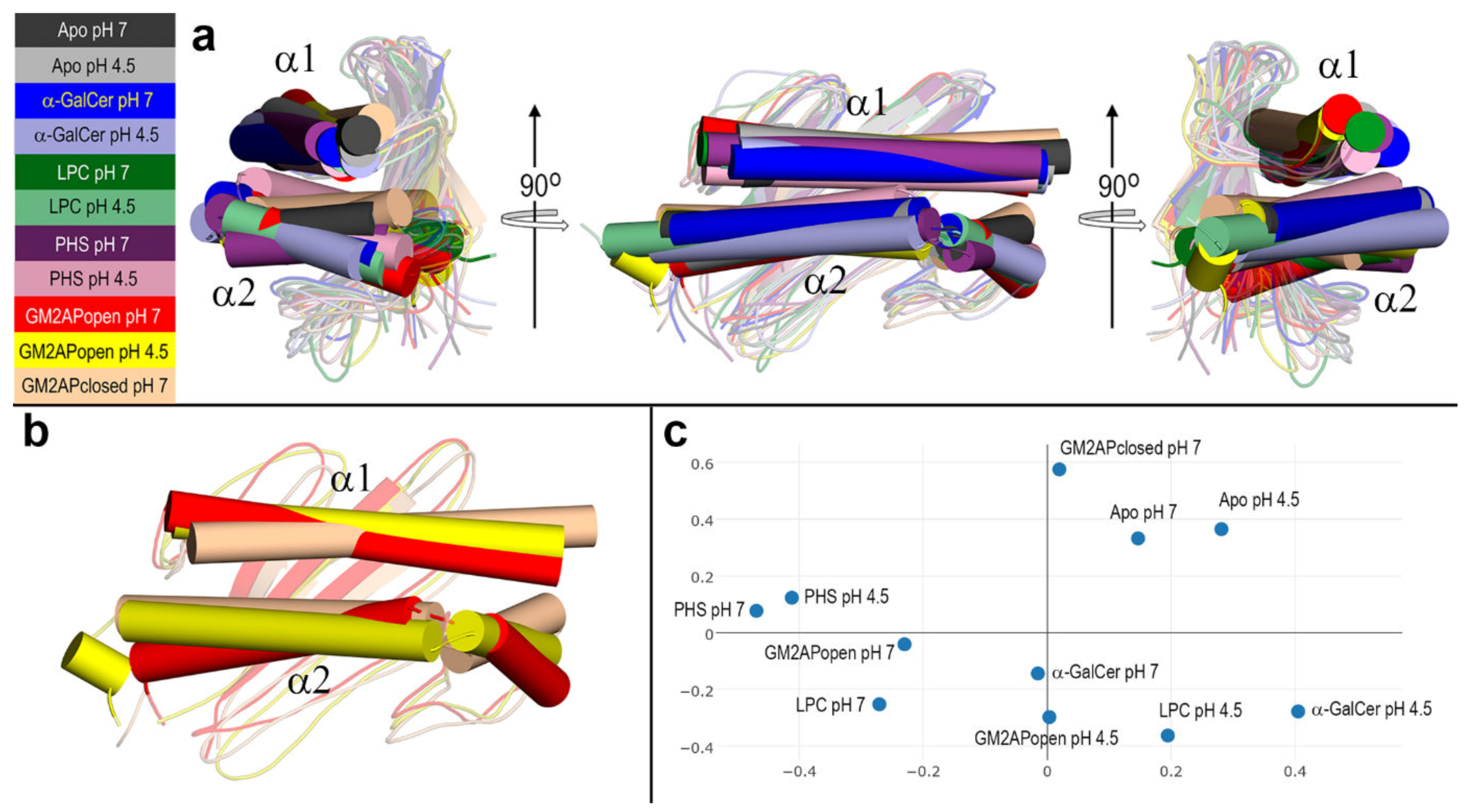
3.4. Final Structures of CD1d after MD Simulations
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzen, A.K.; Such, L.; Jensen, K.K.; Marquard, A.M.; Jessen, L.E.; Miller, N.J.; Church, C.D.; Lyngaa, R.; Koelle, D.M.; Becker, J.C.; et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide–MHC complexes. Nat. Biotechnol. 2018, 36, 1191–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barral, D.C.; Brenner, M.B. CD1 antigen presentation: How it works. Nat. Rev. Immunol. 2007, 7, 929–941. [Google Scholar] [CrossRef]
- Vartabedian, V.F.; Savage, P.B.; Teyton, L. The processing and presentation of lipids and glycolipids to the immune system. Immunol. Rev. 2016, 272, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chancellor, A.; Gadola, S.D.; Mansour, S. The versatility of the CD1 lipid antigen presentation pathway. Immunology 2018, 154, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alles, L.F.; Giacometti, G.; Versluis, C.; Maveyraud, L.; de Paepe, D.; Guiard, J.; Tranier, S.; Gilleron, M.; Prandi, J.; Hanau, D.; et al. Crystal structure of human CD1e reveals a groove suited for lipid-exchange processes. Proc. Natl. Acad. Sci. USA 2011, 108, 13230–13235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardi, E.; Zajonc, D. Molecular basis of lipid antigen presentation by CD1d and recognition by natural killer T cells. Immunol. Rev. 2012, 250, 167–179. [Google Scholar] [CrossRef]
- Wun, K.S.; Cameron, G.; Patel, O.; Pang, S.S.; Pellicci, D.G.; Sullivan, L.C.; Keshipeddy, S.; Young, M.H.; Uldrich, A.P.; Thakur, M.S.; et al. A molecular basis for the exquisite CD1d-restricted antigen specificity and functional responses of natural killer T cells. Immunity 2011, 34, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef]
- Brennan, P.J.; Cheng, T.-Y.; Pellicci, D.G.; Watts, G.F.M.; Veerapen, N.; Young, D.C.; Rossjohn, J.; Besra, G.S.; Godfrey, D.I.; Brenner, M.B.; et al. Structural determination of lipid antigens captured at the CD1d–T-cell receptor interface. Proc. Natl. Acad. Sci. USA 2017, 114, 8348–8353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teyton, L. Role of lipid transfer proteins in loading CD1 antigen-presenting molecules. J. Lipid Res. 2018, 59, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Sagiv, Y.; Benlagha, K.; Bendelac, A.; Cantu, C., III; Schrantz, N.; Teyton, L.; Kulkarni, A.B.; Qi, X.; Grabowski, G.A.; et al. Editing of CD1d-Bound Lipid Antigens by Endosomal Lipid Transfer Proteins. Science 2004, 303, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freigang, S.; Zadorozhny, V.; McKinney, M.K.; Krebs, P.; Herro, R.; Pawlak, J.; Kain, L.; Schrantz, N.; Masuda, K.; Liu, Y.; et al. Fatty acid amide hydrolase shapes NKT cell responses by influencing the serum transport of lipid antigen in mice. J. Clin. Investig. 2010, 120, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Garzón, D.; Anselmi, C.; Bond, P.J.; Faraldo-Gómez, J.D. Dynamics of the Antigen-binding Grooves in CD1 Proteins. J. Biol. Chem. 2013, 288, 19528–19536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, S.; Tocheva, A.S.; Cave-Ayland, C.; Machelett, M.M.; Sander, B.; Lissin, N.M.; Molloy, P.E.; Baird, M.S.; Stübs, G.; Schröder, N.W.J.; et al. Cholesteryl esters stabilize human CD1c conformations for recognition by self-reactive T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E1266–E1275. [Google Scholar] [CrossRef] [Green Version]
- Chancellor, A.; Tocheva, A.S.; Cave-Ayland, C.; Tezera, L.; White, A.; Al Dulayymi, J.R.; Bridgeman, J.S.; Tews, I.; Wilson, S.; Lissin, N.M.; et al. CD1b-restricted GEM T cell responses are modulated by Mycobacterium tuberculosis mycolic acid meromycolate chains. Proc. Natl. Acad. Sci. USA 2017, 114, E10956–E10964. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; Gatta, A.T.; Levine, T.P. Lipid transfer proteins: The lipid commute via shuttles, bridges and tubes. Nat. Rev. Mol. Cell Biol. 2019, 20, 85–101. [Google Scholar] [CrossRef]
- López-Sagaseta, J.; Sibener, L.V.; Kung, J.E.; Gumperz, J.; Adams, E.J. Lysophospholipid presentation by CD1d and recognition by a human Natural Killer T-cell receptor. EMBO J. 2012, 31, 2047–2059. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.T.; Huang, J.R.; Yu, A.L. Tailored design of NKT-stimulatory glycolipids for polarization of immune responses. J. Biomed. Sci. 2017, 24, 22. [Google Scholar] [CrossRef] [Green Version]
- Dangerfield, E.M.; Cheng, J.M.H.; Knight, D.A.; Weinkove, R.; Dunbar, P.R.; Hermans, I.F.; Timmer, M.S.M.; Stocker, B.L. Species-Specific Activity of Glycolipid Ligands for Invariant NKT Cells. ChemBioChem 2012, 13, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Cubells-Baeza, N.; Gómez-Casado, C.; Tordesillas, L.; Ramírez-Castillejo, C.; Garrido-Arandia, M.; González-Melendi, P.; Herrero, M.; Pacios, L.F.; Díaz-Perales, A. Identification of the ligand of Pru p 3, a peach LTP. Plant Mol. Biol. 2017, 94, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordesillas, L.; Cubells-Baeza, N.; Gómez-Casado, C.; Berin, C.; Esteban, V.; Barcik, W.; O’Mahony, L.; Ramirez, C.; Pacios, L.F.; Garrido-Arandia, M.; et al. Mechanisms underlying induction of allergic sensitization by Pru p 3. Clin. Exp. Allergy 2017, 47, 1398–1408. [Google Scholar] [CrossRef]
- Koch, M.; Stronge, V.S.; Shepherd, D.; Gadola, S.D.; Mathew, B.; Ritter, G.; Fersht, A.R.; Besra, G.S.; Schmidt, R.R.; Jones, E.Y.; et al. The crystal structure of human CD1d with and without α-galactosylceramide. Nat. Immunol. 2005, 6, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Zuviría, B.; Garrido-Arandia, M.; Díaz-Perales, A.; Pacios, L.F. Energy landscapes of ligand motion inside the Tunnel-like cavity of lipid transfer proteins: The case of the Pru p 3 allergen. Int. J. Mol. Sci. 2019, 20, 1432. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.S.; Mi, L.Z.; Lee, S.; Rastinejad, F. Crystal structure analysis of phosphatidylcholine―GM2-activator product complexes: Evidence for hydrolase activity. Biochemistry 2005, 44, 13510–13521. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. Autodock vina: Improving the speed and accuracy of docking. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera―A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Jiménez-García, B.; Pons, C.; Fernández-Recio, J. pyDockWEB: A web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [Green Version]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Lyskov, S.; Gray, J.J. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008, 36, W233–W238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.H.M.; SØndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins Struct. Funct. Genet. 2006, 63, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilambi, K.P.; Gray, J.J. Rapid calculation of protein pKa values using rosetta. Biophys. J. 2012, 103, 587–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, LLC. The PyMol Molecular Graphics System, Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2019. [Google Scholar]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Koukos, P.I.; Glykos, N.M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Modeling 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Holm, L.; Park, J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. [Google Scholar] [CrossRef]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Dalchau, N.; Carter, R.; Emmott, S.; Phillips, A.; Werner, J.M.; Elliott, T. Selector function of MHC I molecules is determined by protein plasticity. Sci. Rep. 2015, 5, 14928. [Google Scholar] [CrossRef] [PubMed]
- Van Hateren, A.; Anderson, M.; Bailey, A.; Werner, J.M.; Skipp, P.; Elliott, T. Direct evidence for conformational dynamics in major histocompatibility complex class I molecules. J. Biol. Chem. 2017, 292, 20255–20269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, Y.; Fukamizo, T.; Okajima, T.; Goto, S.; Boucher, I.; Brzezinski, R. Thermal unfolding of chitosanase from Streptomyces sp. N174: Role of tryptophan residues in the protein structure stabilization. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1999, 1429, 365–376. [Google Scholar] [CrossRef]
- Wallace, L.A.; Burke, J.; Dirr, H.W. Domain-domain interface packing at conserved Trp-20 in class α glutathione transferase impacts on protein stability. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1478, 325–332. [Google Scholar] [CrossRef]
- Huntley, J.J.A.; Fast, W.; Benkovic, S.J.; Wright, P.E.; Dyson, H.J. Role of a solvent-exposed tryptophan in the recognition and binding of antibiotic substrates for a metallo-β-lactamase. Protein Sci. 2003, 12, 1368–1375. [Google Scholar] [CrossRef]
- Campanini, B.; Raboni, S.; Vaccari, S.; Zhang, L.; Cook, P.F.; Hazlett, T.L.; Mozzarelli, A.; Bettati, S. Surface-exposed tryptophan residues are essential for O-acetylserine sulfhydrylase structure, function, and stability. J. Biol. Chem. 2003, 278, 37511–37519. [Google Scholar] [CrossRef] [Green Version]
- Karadi, K.; Kapetanaki, S.M.; Raics, K.; Pecsi, I.; Kapronczai, R.; Fekete, Z.; Iuliano, J.N.; Collado, J.T.; Gil, A.A.; Orban, J.; et al. Functional dynamics of a single tryptophan residue in a BLUF protein revealed by fluorescence spectroscopy. Sci. Rep. 2020, 10, 2061. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Pellicci, D.G.; Cheng, T.Y.; Keller, A.N.; Sandoval-Romero, M.; Gras, S.; De Jong, A.; Uldrich, A.P.; Moody, D.B.; Godfrey, D.I.; et al. αβ T cell antigen receptor recognition of CD1a presenting self lipid ligands. Nat. Immunol. 2015, 16, 258–266. [Google Scholar] [CrossRef]
- Shahine, A.; Reinink, P.; Reijneveld, J.F.; Gras, S.; Holzheimer, M.; Cheng, T.Y.; Minnaard, A.J.; Altman, J.D.; Lenz, S.; Prandi, J.; et al. A T-cell receptor escape channel allows broad T-cell response to CD1b and membrane phospholipids. Nat. Commun. 2019, 10, 56. [Google Scholar] [CrossRef]
- Wun, K.S.; Reijneveld, J.F.; Cheng, T.Y.; Ladell, K.; Uldrich, A.P.; Le Nours, J.; Miners, K.L.; McLaren, J.E.; Grant, E.J.; Haigh, O.L.; et al. T cell autoreactivity directed toward CD1c itself rather than toward carried self lipids. Nat. Immunol. 2018, 19, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Paletta, D.; Fichtner, A.S.; Starick, L.; Porcelli, S.A.; Savage, P.B.; Herrmann, T. Species specific differences of CD1d oligomer loading in vitro. PLoS ONE 2015, 10, 0143449. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD1d System | Trp140 | Trp153 | Trp160 |
|---|---|---|---|
| Apo form pH 7 | 58.4 (12.1) | 173.8 (4.8) | 167.4 (17.7) |
| Apo for pH 4.5 | 69.0 (16.6) | 173.0 (5.3) | 172.2 (6.1) |
| α-GalCer pH 7 | 57.3 (11.6) | 170.9 (6.5) | 169.9 (7.4) |
| α-GalCer pH 4.5 | 52.7 (10.9) | 172.7 (5.4) | 170.7 (6.4) |
| LPC pH 7 | 56.4 (11.0) | −47.1 (50.8) | 170.5 (7.0) |
| LPC pH 4.5 | 55.8 (11.4) | 133.3 (87.0) | 171.2 (6.8) |
| PHS pH 7 | 46.1 (10.1) | 171.8 (6.2) | 170.6 (6.6) |
| PHS pH 4.5 | 16.0 (49.4) | 172.6 (5.8) | 170.5 (7.3) |
| GM2APopen pH 7 | 60.4 (12.5) | −59.5 (11.7) | 169.7 (7.8) |
| GM2APopen pH 4.5 | 56.2 (12.8) | 163.4 (44.9) | 171.4 (6.9) |
| GM2APclosed pH 7 | 70.9 (11.2) | 173.3 (5.3) | 170.6 (7.1) |
| CD1d System | α1 | α2 |
|---|---|---|
| Apo form pH 7 | 60‒88 | 139‒150 + 153‒177 |
| Apo for pH 4.5 | 60‒88 | 139‒150 + 152‒181 |
| α-GalCer pH 7 | 60‒88 | 139‒150 + 152‒176 |
| α-GalCer pH 4.5 | 60‒88 | 139‒149 + 152‒181 |
| LPC pH 7 | 60‒88 | 139‒150 + 153‒181 |
| LPC pH 4.5 | 60‒88 | 139‒150 + 153‒182 |
| PHS pH 7 | 60‒88 | 139‒150 + 152‒177 |
| PHS pH 4.5 | 60‒88 | 139‒149 + 153‒176 |
| GM2APopen pH 7 | 60‒88 | 139‒149 + 153‒182 |
| GM2APopen pH 4.5 | 60‒88 | 139‒150 + 152‒176 + 179‒181 |
| GM2APclosed pH 7 | 60‒88 | 139‒150 + 152‒178 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas-Zuviría, B.; Mínguez-Toral, M.; Díaz-Perales, A.; Garrido-Arandia, M.; Pacios, L.F. Structural Dynamics of the Lipid Antigen-Binding Site of CD1d Protein. Biomolecules 2020, 10, 532. https://doi.org/10.3390/biom10040532
Cuevas-Zuviría B, Mínguez-Toral M, Díaz-Perales A, Garrido-Arandia M, Pacios LF. Structural Dynamics of the Lipid Antigen-Binding Site of CD1d Protein. Biomolecules. 2020; 10(4):532. https://doi.org/10.3390/biom10040532
Chicago/Turabian StyleCuevas-Zuviría, Bruno, Marina Mínguez-Toral, Araceli Díaz-Perales, María Garrido-Arandia, and Luis F. Pacios. 2020. "Structural Dynamics of the Lipid Antigen-Binding Site of CD1d Protein" Biomolecules 10, no. 4: 532. https://doi.org/10.3390/biom10040532
APA StyleCuevas-Zuviría, B., Mínguez-Toral, M., Díaz-Perales, A., Garrido-Arandia, M., & Pacios, L. F. (2020). Structural Dynamics of the Lipid Antigen-Binding Site of CD1d Protein. Biomolecules, 10(4), 532. https://doi.org/10.3390/biom10040532