ADAMTS-15 Has a Tumor Suppressor Role in Prostate Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Prostate Cancer Samples
2.2. Immunofluorescence
2.3. Cell Culture and Transfection
2.4. Western Blot
2.5. Migration Assay
2.6. Proliferation Assay
2.7. Apoptosis Assay
2.8. Animal Methods
2.9. Tumor Analysis
2.10. Statistical Analysis
3. Results
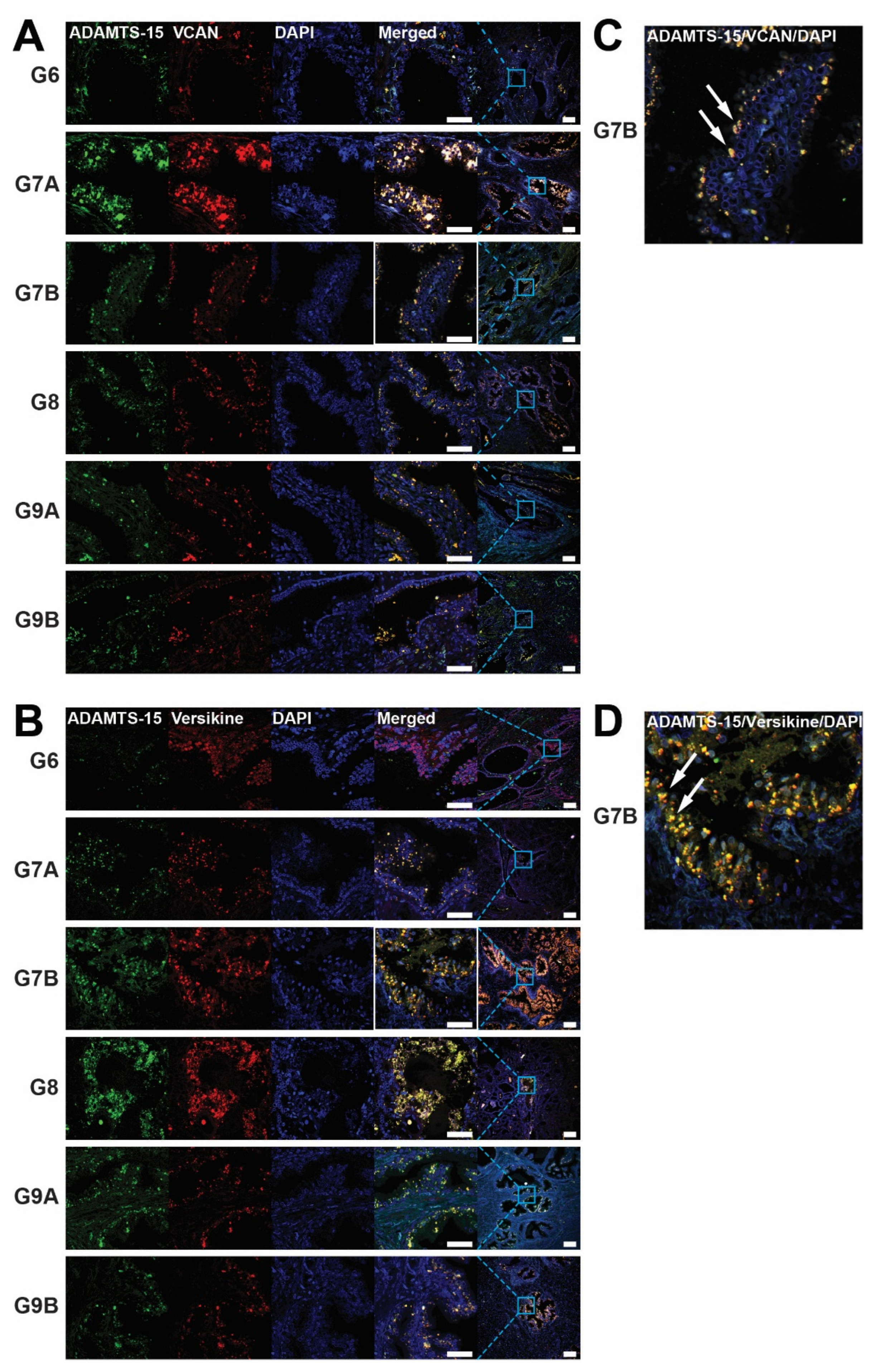
3.1. Localization of ADAMTS-15, VCAN and Versikine in Human Prostate Cancer Biopsies
3.2. Enforced ADAMTS-15 Expression in Prostate Cancer Cell Lines
3.3. Effect of ADAMTS-15 Expression on Proliferation
3.4. Effect of ADAMTS-15 Expression on Survival
3.5. Effect of ADAMTS-15 Expression on Migration
3.6. Effect of ADAMTS-15 Expression on Androgen-Responsive Tumor Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. Ca Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, J.T.; Isaacs, J.T. Mechanisms involved in the progression of androgen-independent prostate cancers: It is not only the cancer cell’s fault. Endocr. Relat. Cancer 2002, 9, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Schopfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.S.; Gao, A.C. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- de Kruijf, E.M.; van Nes, J.G.; van de Velde, C.J.; Putter, H.; Smit, V.T.; Liefers, G.J.; Kuppen, P.J.; Tollenaar, R.A.; Mesker, W.E. Tumor-stroma ratio in the primary tumor is a prognostic factor in early breast cancer patients, especially in triple-negative carcinoma patients. Breast Cancer Res. Treat. 2011, 125, 687–696. [Google Scholar] [CrossRef]
- Suhovskih, A.V.; Mostovich, L.A.; Kunin, I.S.; Boboev, M.M.; Nepomnyashchikh, G.I.; Aidagulova, S.V.; Grigorieva, E.V. Proteoglycan expression in normal human prostate tissue and prostate cancer. Isrn Oncol. 2013, 2013, 680136. [Google Scholar] [CrossRef]
- Januchowski, R.; Zawierucha, P.; Rucinski, M.; Nowicki, M.; Zabel, M. Extracellular matrix proteins expression profiling in chemoresistant variants of the A2780 ovarian cancer cell line. Biomed. Res. Int. 2014, 2014, 365867. [Google Scholar] [CrossRef] [Green Version]
- Blehm, B.H.; Jiang, N.; Kotobuki, Y.; Tanner, K. Deconstructing the role of the ECM microenvironment on drug efficacy targeting MAPK signaling in a pre-clinical platform for cutaneous melanoma. Biomaterials 2015, 56, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Fullar, A.; Dudas, J.; Olah, L.; Hollosi, P.; Papp, Z.; Sobel, G.; Karaszi, K.; Paku, S.; Baghy, K.; Kovalszky, I. Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. Bmc Cancer 2015, 15, 256. [Google Scholar] [CrossRef] [Green Version]
- Suhovskih, A.V.; Aidagulova, S.V.; Kashuba, V.I.; Grigorieva, E.V. Proteoglycans as potential microenvironmental biomarkers for colon cancer. Cell Tissue Res. 2015, 361, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the regulation of cell phenotype in disease. Biochim. Biophys. Acta 2014, 1840, 2441–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakko, A.J.; Ricciardelli, C.; Mayne, K.; Tilley, W.D.; Lebaron, R.G.; Horsfall, D.J. Versican accumulation in human prostatic fibroblast cultures is enhanced by prostate cancer cell-derived transforming growth factor beta1. Cancer Res. 2001, 61, 926–930. [Google Scholar] [PubMed]
- Sakko, A.J.; Ricciardelli, C.; Mayne, K.; Suwiwat, S.; LeBaron, R.G.; Marshall, V.R.; Tilley, W.D.; Horsfall, D.J. Modulation of prostate cancer cell attachment to matrix by versican. Cancer Res. 2003, 63, 4786–4791. [Google Scholar]
- Ricciardelli, C.; Russell, D.L.; Ween, M.P.; Mayne, K.; Suwiwat, S.; Byers, S.; Marshall, V.R.; Tilley, W.D.; Horsfall, D.J. Formation of hyaluronan- and versican-rich pericellular matrix by prostate cancer cells promotes cell motility. J. Biol. Chem. 2007, 282, 10814–10825. [Google Scholar] [CrossRef] [Green Version]
- Dancevic, C.M.; Fraser, F.W.; Smith, A.D.; Stupka, N.; Ward, A.C.; McCulloch, D.R. The biosynthesis and expression of A Disintegrin-like and Metalloproteinase domain with Thrombospondin-1 repeats -15: A novel versican-cleaving proteoglycanase. J. Biol. Chem. 2013, 288, 37267–37276. [Google Scholar] [CrossRef] [Green Version]
- Read, J.T.; Rahmani, M.; Boroomand, S.; Allahverdian, S.; McManus, B.M.; Rennie, P.S. Androgen receptor regulation of the versican gene through an androgen response element in the proximal promoter. J. Biol. Chem. 2007, 282, 31954–31963. [Google Scholar] [CrossRef] [Green Version]
- Sandy, J.D.; Westling, J.; Kenagy, R.D.; Iruela-Arispe, M.L.; Verscharen, C.; Rodriguez-Mazaneque, J.C.; Zimmermann, D.R.; Lemire, J.M.; Fischer, J.W.; Wight, T.N.; et al. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J. Biol. Chem. 2001, 276, 13372–13378. [Google Scholar] [CrossRef] [Green Version]
- Somerville, R.P.; Longpre, J.M.; Jungers, K.A.; Engle, J.M.; Ross, M.; Evanko, S.; Wight, T.N.; Leduc, R.; Apte, S.S. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J. Biol. Chem. 2003, 278, 9503–9513. [Google Scholar] [CrossRef] [Green Version]
- Longpre, J.M.; McCulloch, D.R.; Koo, B.H.; Alexander, J.P.; Apte, S.S.; Leduc, R. Characterization of proADAMTS5 processing by proprotein convertases. Int. J. Biochem. Cell Biol. 2009, 41, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.R.; Nelson, C.M.; Dixon, L.J.; Silver, D.L.; Wylie, J.D.; Lindner, V.; Sasaki, T.; Cooley, M.A.; Argraves, W.S.; Apte, S.S. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev. Cell 2009, 17, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandadasa, S.; Foulcer, S.; Apte, S.S. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014, 35, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 2016, 128, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.J.; McCoombe, S.; Williams, E.D.; McCulloch, D.R.; Ward, A.C. The extracellular matrix in cancer progression: Role of hyalectan proteoglycans and ADAMTS enzymes. Cancer Lett. 2017, 385, 55–64. [Google Scholar] [CrossRef]
- Cross, N.A.; Chandrasekharan, S.; Jokonya, N.; Fowles, A.; Hamdy, F.C.; Buttle, D.J.; Eaton, C.L. The expression and regulation of ADAMTS-1, -4, -5, -9, and -15, and TIMP-3 by TGFbeta1 in prostate cells: Relevance to the accumulation of versican. Prostate 2005, 63, 269–275. [Google Scholar] [CrossRef]
- Gustavsson, H.; Wang, W.; Jennbacken, K.; Welen, K.; Damber, J.E. ADAMTS1, a putative anti-angiogenic factor, is decreased in human prostate cancer. BJU Int. 2009, 104, 1786–1790. [Google Scholar] [CrossRef]
- Molokwu, C.N.; Adeniji, O.O.; Chandrasekharan, S.; Hamdy, F.C.; Buttle, D.J. Androgen regulates ADAMTS15 gene expression in prostate cancer cells. Cancer Invest. 2010, 28, 698–710. [Google Scholar] [CrossRef]
- Porter, S.; Span, P.N.; Sweep, F.C.; Tjan-Heijnen, V.C.; Pennington, C.J.; Pedersen, T.X.; Johnsen, M.; Lund, L.R.; Romer, J.; Edwards, D.R. ADAMTS8 and ADAMTS15 expression predicts survival in human breast carcinoma. Int. J. Cancer 2006, 118, 1241–1247. [Google Scholar] [CrossRef]
- Viloria, C.G.; Obaya, A.J.; Moncada-Pazos, A.; Llamazares, M.; Astudillo, A.; Capella, G.; Cal, S.; Lopez-Otin, C. Genetic inactivation of ADAMTS15 metalloprotease in human colorectal cancer. Cancer Res. 2009, 69, 4926–4934. [Google Scholar] [CrossRef] [Green Version]
- Kelwick, R.; Wagstaff, L.; Decock, J.; Roghi, C.; Cooley, L.S.; Robinson, S.D.; Arnold, H.; Gavrilovic, J.; Jaworski, D.M.; Yamamoto, K.; et al. Metalloproteinase-dependent and -independent processes contribute to inhibition of breast cancer cell migration, angiogenesis and liver metastasis by a disintegrin and metalloproteinase with thrombospondin motifs-15. Int. J. Cancer 2015, 136, E14–E26. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Fraser, F.W.; Riley, C.; Ahmed, N.; McCulloch, D.R.; Ward, A.C. Granulocyte colony-stimulating factor receptor signalling via Janus kinase 2/Signal transducer and activator of transcription 3 in ovarian cancer. Br. J. Cancer 2014, 110, 133–145. [Google Scholar] [CrossRef]
- Jensen, E.C. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat. Rec. 2013, 296, 378–381. [Google Scholar] [CrossRef]
- Li, Q.; Lau, A.; Morris, T.J.; Guo, L.; Fordyce, C.B.; Stanley, E.F. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: Analysis by quantitative immunocolocalization. J. Neurosci. 2004, 24, 4070–4081. [Google Scholar] [CrossRef] [Green Version]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Bastide, C.; Bagnis, C.; Mannoni, P.; Hassoun, J.; Bladou, F. A Nod Scid mouse model to study human prostate cancer. Prostate Cancer Prostatic Dis. 2002, 5, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancevic, C.M.; McCulloch, D.R.; Ward, A.C. The ADAMTS hyalectanase family: Biological insights from diverse species. Biochem. J. 2016, 473, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- El Hour, M.; Moncada-Pazos, A.; Blacher, S.; Masset, A.; Cal, S.; Berndt, S.; Detilleux, J.; Host, L.; Obaya, A.J.; Maillard, C.; et al. Higher sensitivity of Adamts12-deficient mice to tumor growth and angiogenesis. Oncogene 2010, 29, 3025–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Sharghi-Namini, S.; Rao, N.; Ge, R. ADAMTS5 functions as an anti-angiogenic and anti-tumorigenic protein independent of its proteoglycanase activity. Am. J. Pathol. 2012, 181, 1056–1068. [Google Scholar] [CrossRef]
- Wagstaff, L.; Kelwick, R.; Decock, J.; Edwards, D.R. The roles of ADAMTS metalloproteinases in tumorigenesis and metastasis. Front. Biosci. 2011, 16, 1861–1872. [Google Scholar] [CrossRef] [Green Version]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [Green Version]
- Loeb, S.; Folkvaljon, Y.; Robinson, D.; Lissbrant, I.F.; Egevad, L.; Stattin, P. Evaluation of the 2015 Gleason grade groups in a nationwide population-based cohort. Eur. Urol. 2016, 69, 1135–1141. [Google Scholar] [CrossRef] [Green Version]
- Nagyova, E.; Salustri, A.; Nemcova, L.; Scsukova, S.; Kalous, J.; Camaioni, A. Versican G1 fragment establishes a strongly stabilized interaction with hyaluronan-rich expanding matrix during oocyte maturation. Int. J. Mol. Sci. 2020, 21, 2267. [Google Scholar] [CrossRef] [Green Version]
- Datta, M.W.; Hernandez, A.M.; Schlicht, M.J.; Kahler, A.J.; DeGueme, A.M.; Dhir, R.; Shah, R.B.; Farach-Carson, C.; Barrett, A.; Datta, S. Perlecan, a candidate gene for the CAPB locus, regulates prostate cancer cell growth via the Sonic Hedgehog pathway. Mol. Cancer 2006, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.; Misra, S.; Toole, B.P. Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J. Biol. Chem. 2002, 277, 38013–38020. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, I.E.; Culig, Z.; Ramoner, R.; Thurnher, M.; Putz, T.; Nessler-Menardi, C.; Tiefenthaler, M.; Bartsch, G.; Klocker, H. Inhibition of LncaP prostate cancer cells by means of androgen receptor antisense oligonucleotides. Cancer Gene. 2000, 7, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltering, K.K.; Helenius, M.A.; Sahu, B.; Manni, V.; Linja, M.J.; Janne, O.A.; Visakorpi, T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009, 69, 8141–8149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, M.; Cernera, G.; Migliaccio, A.; Castoria, G. Nerve growth factor induces proliferation and aggressiveness in prostate cancer cells. Cancers 2019, 11, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, F.; Kraft, J.; Schroeder, C.; Hube-Magg, C.; Kluth, M.; Lang, D.S.; Simon, R.; Sauter, G.; Izbicki, J.R.; Clauditz, T.S.; et al. Up-regulation of biglycan is associated with poor prognosis and PTEN deletion in patients with prostate cancer. Neoplasia 2017, 19, 707–715. [Google Scholar] [CrossRef]
- Sharpe, B.; Alghezi, D.A.; Cattermole, C.; Beresford, M.; Bowen, R.; Mitchard, J.; Chalmers, A.D. A subset of high Gleason grade prostate carcinomas contain a large burden of prostate cancer syndecan-1 positive stromal cells. Prostate 2017, 77, 1312–1324. [Google Scholar] [CrossRef]
- Stupka, N.; Kintakas, C.; White, J.D.; Fraser, F.W.; Hanciu, M.; Aramaki-Hattori, N.; Martin, S.; Coles, C.; Collier, F.; Ward, A.C.; et al. Versican processing by a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats proteinases-5 and -15 facilitates myoblast fusion. J. Biol. Chem. 2013, 288, 1907–1917. [Google Scholar] [CrossRef] [Green Version]
- Arichi, N.; Mitsui, Y.; Hiraki, M.; Nakamura, S.; Hiraoka, T.; Sumura, M.; Hirata, H.; Tanaka, Y.; Dahiya, R.; Yasumoto, H.; et al. Versican is a potential therapeutic target in docetaxel-resistant prostate cancer. Oncoscience 2015, 2, 193–204. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binder, M.J.; McCoombe, S.; Williams, E.D.; McCulloch, D.R.; Ward, A.C. ADAMTS-15 Has a Tumor Suppressor Role in Prostate Cancer. Biomolecules 2020, 10, 682. https://doi.org/10.3390/biom10050682
Binder MJ, McCoombe S, Williams ED, McCulloch DR, Ward AC. ADAMTS-15 Has a Tumor Suppressor Role in Prostate Cancer. Biomolecules. 2020; 10(5):682. https://doi.org/10.3390/biom10050682
Chicago/Turabian StyleBinder, Marley J., Scott McCoombe, Elizabeth D. Williams, Daniel R. McCulloch, and Alister C. Ward. 2020. "ADAMTS-15 Has a Tumor Suppressor Role in Prostate Cancer" Biomolecules 10, no. 5: 682. https://doi.org/10.3390/biom10050682