Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18

 , , , and
, , , and
Abstract
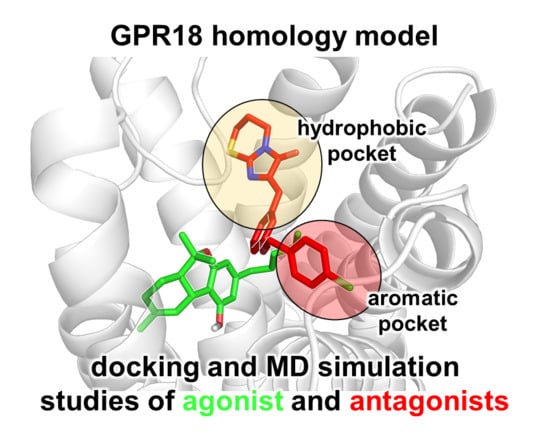
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Homology Modeling
2.2. Docking Studies
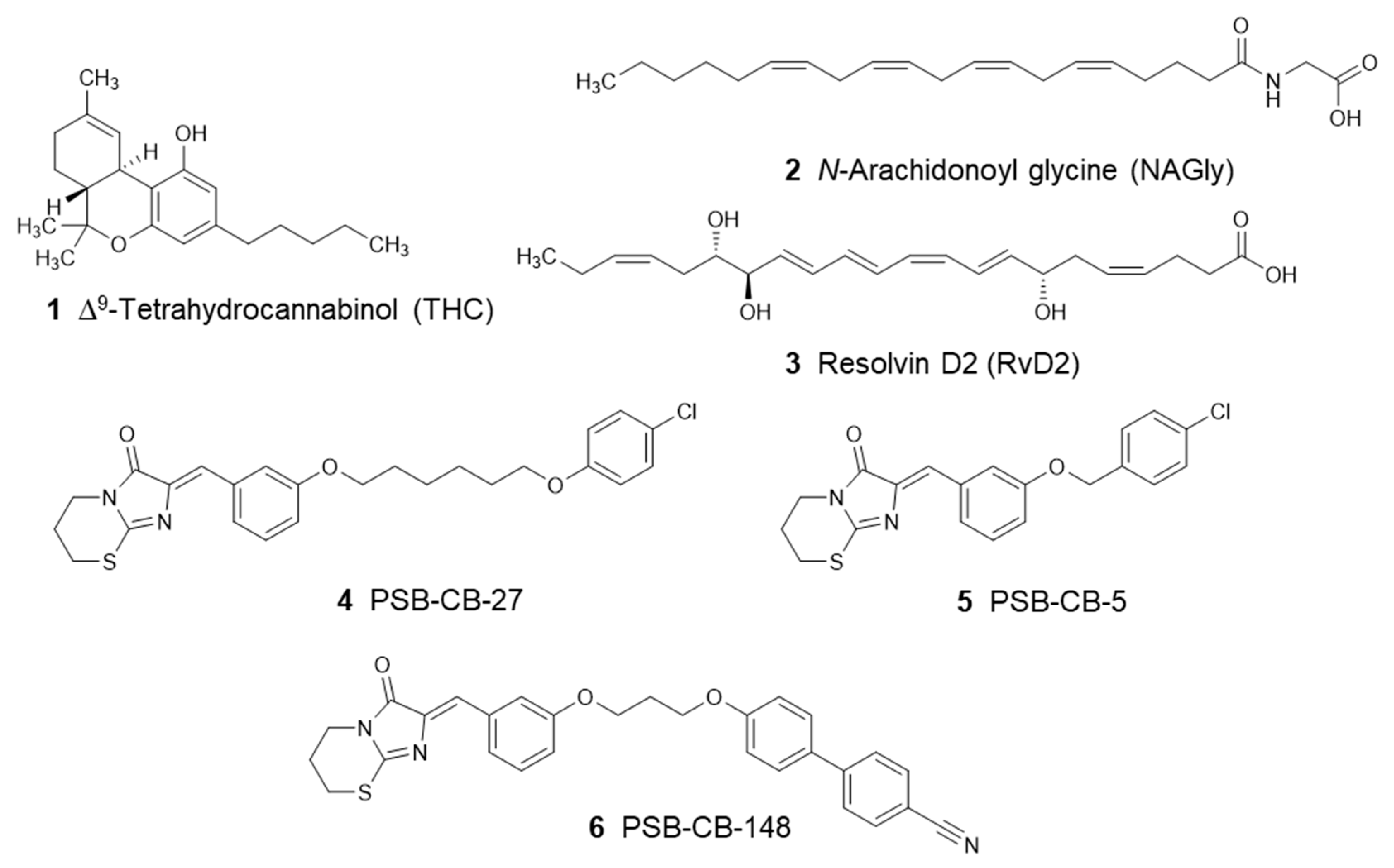
2.3. Compounds
2.4. Molecular Dynamics Simulation
3. Results
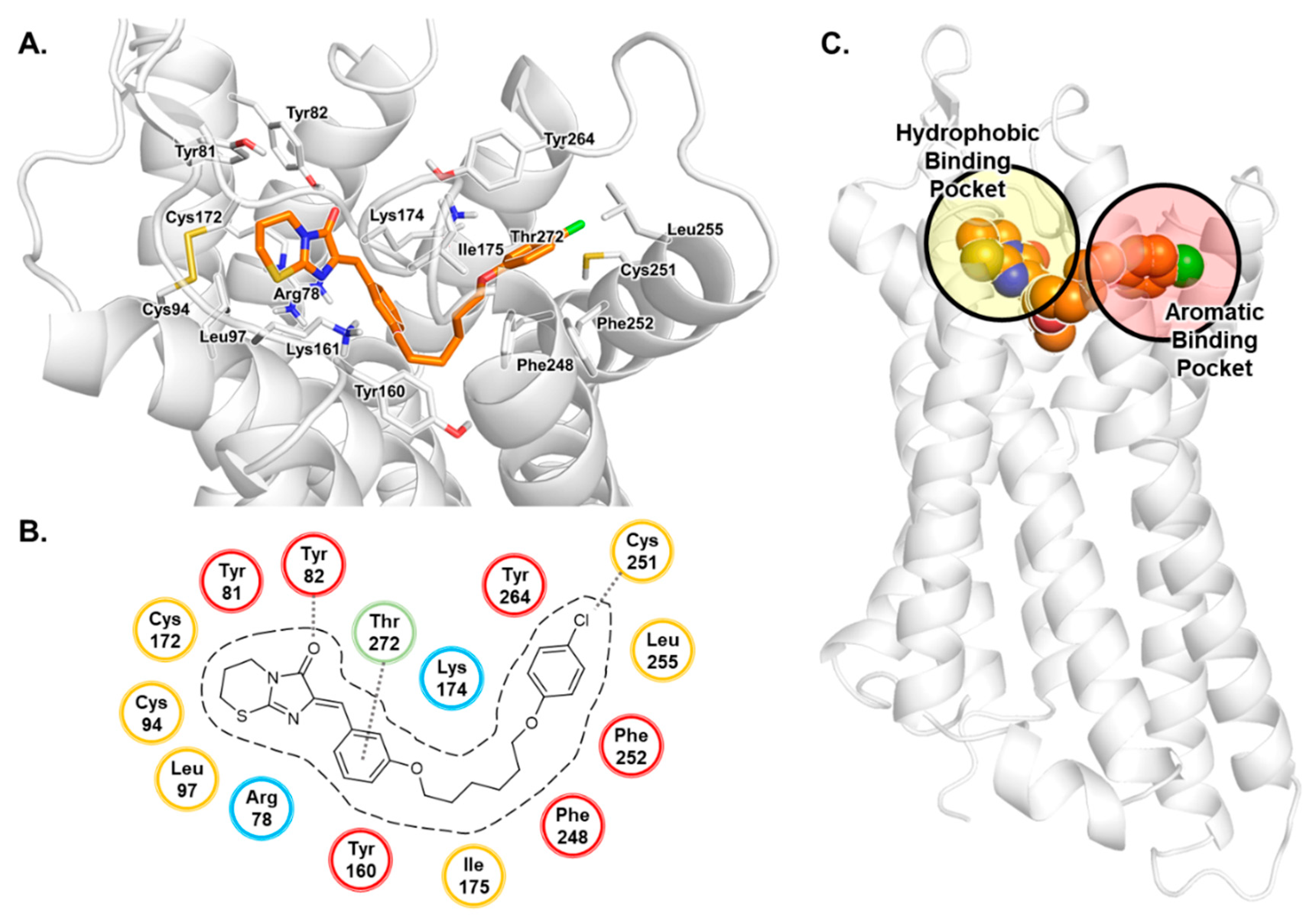
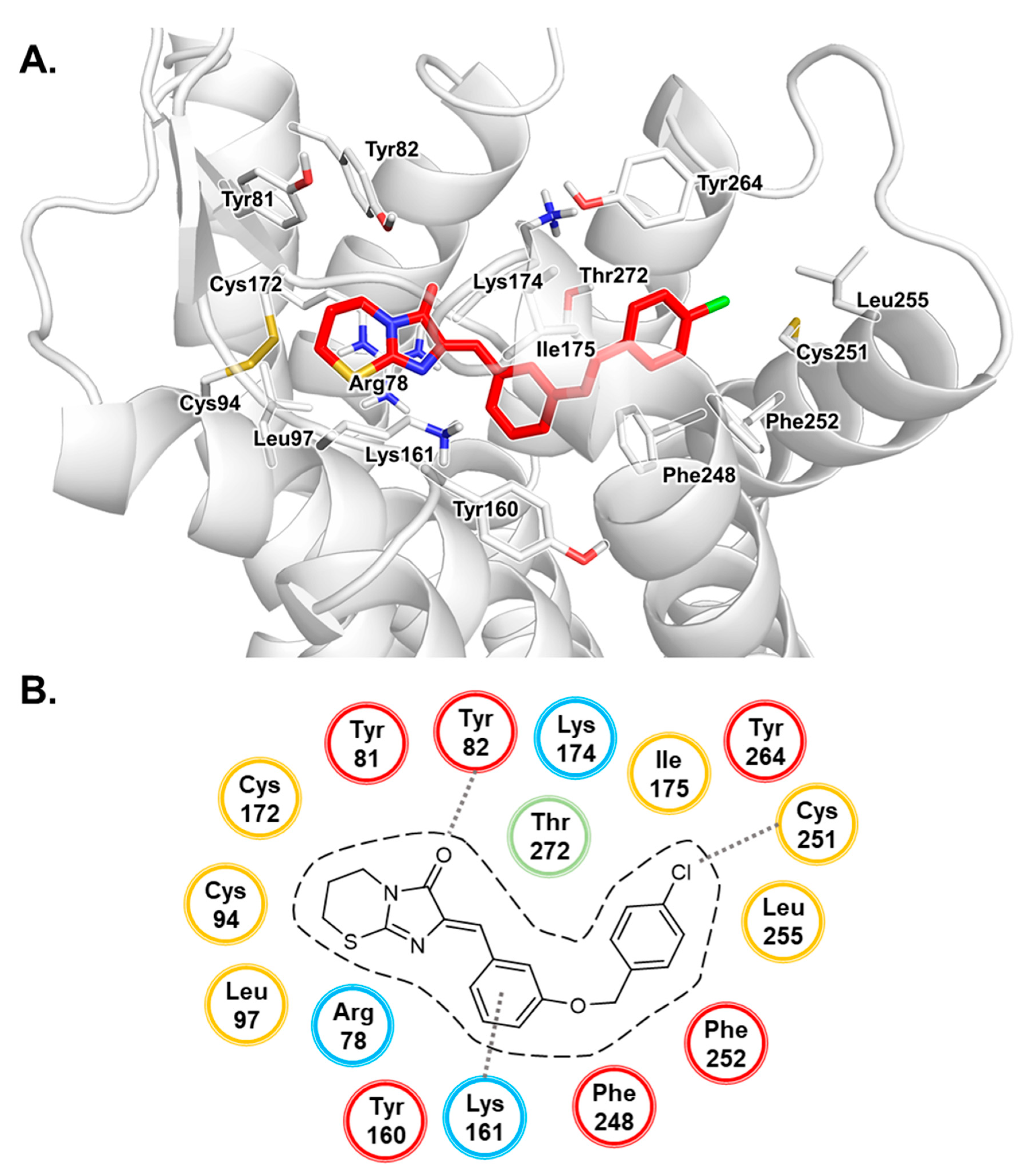
3.1. Docking Studies of Antagonists
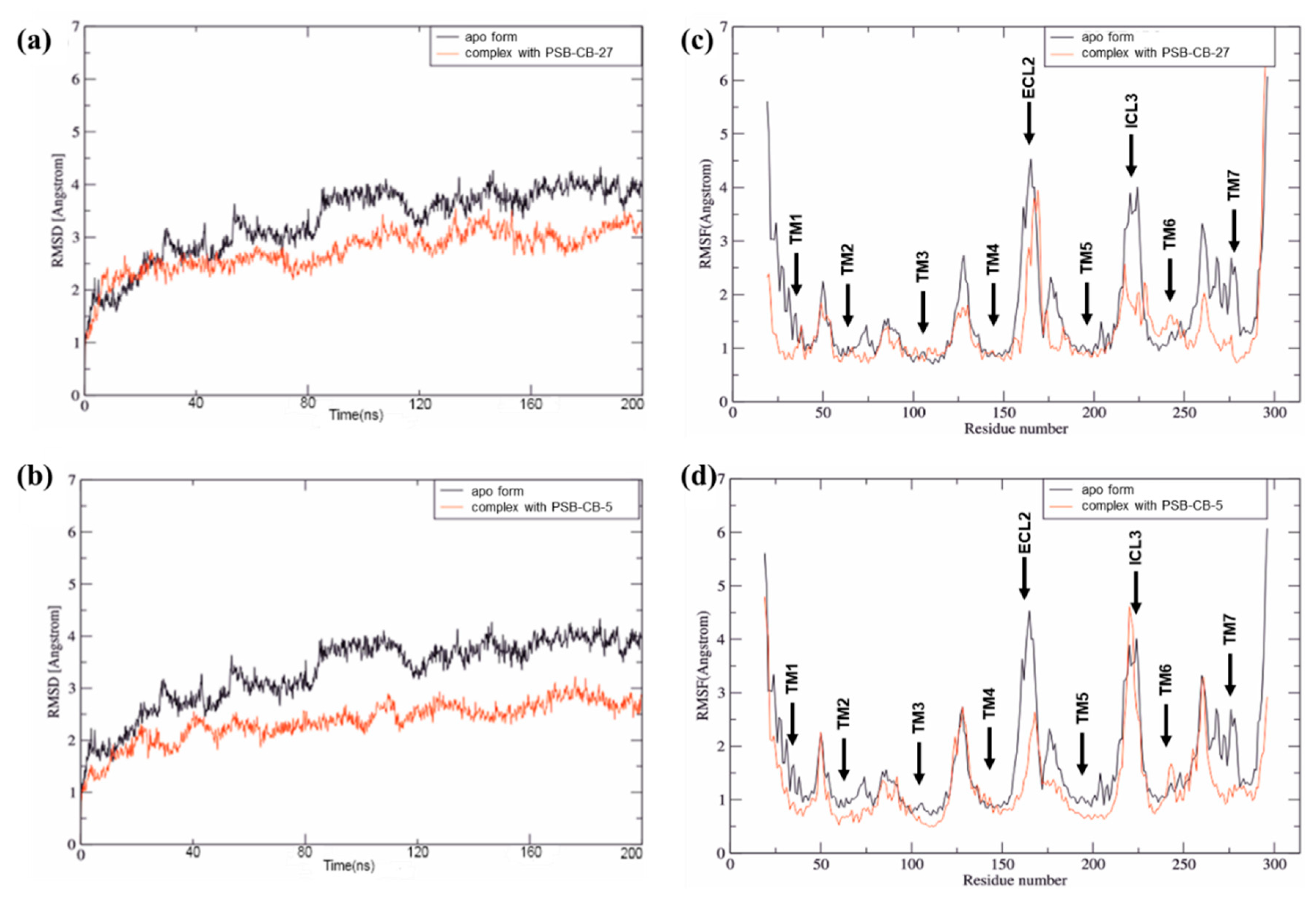
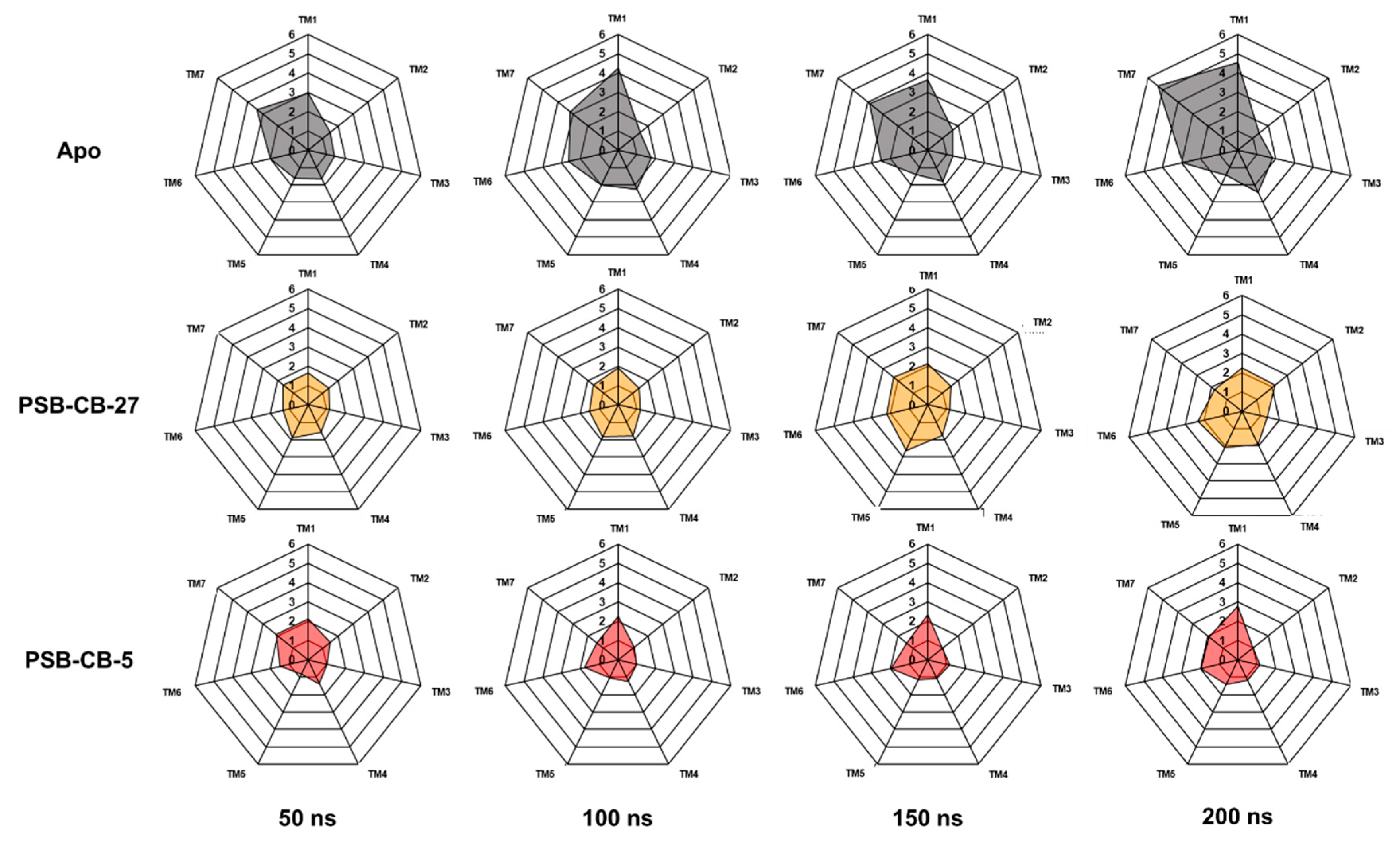
3.2. MD Simulation Study of Antagonists
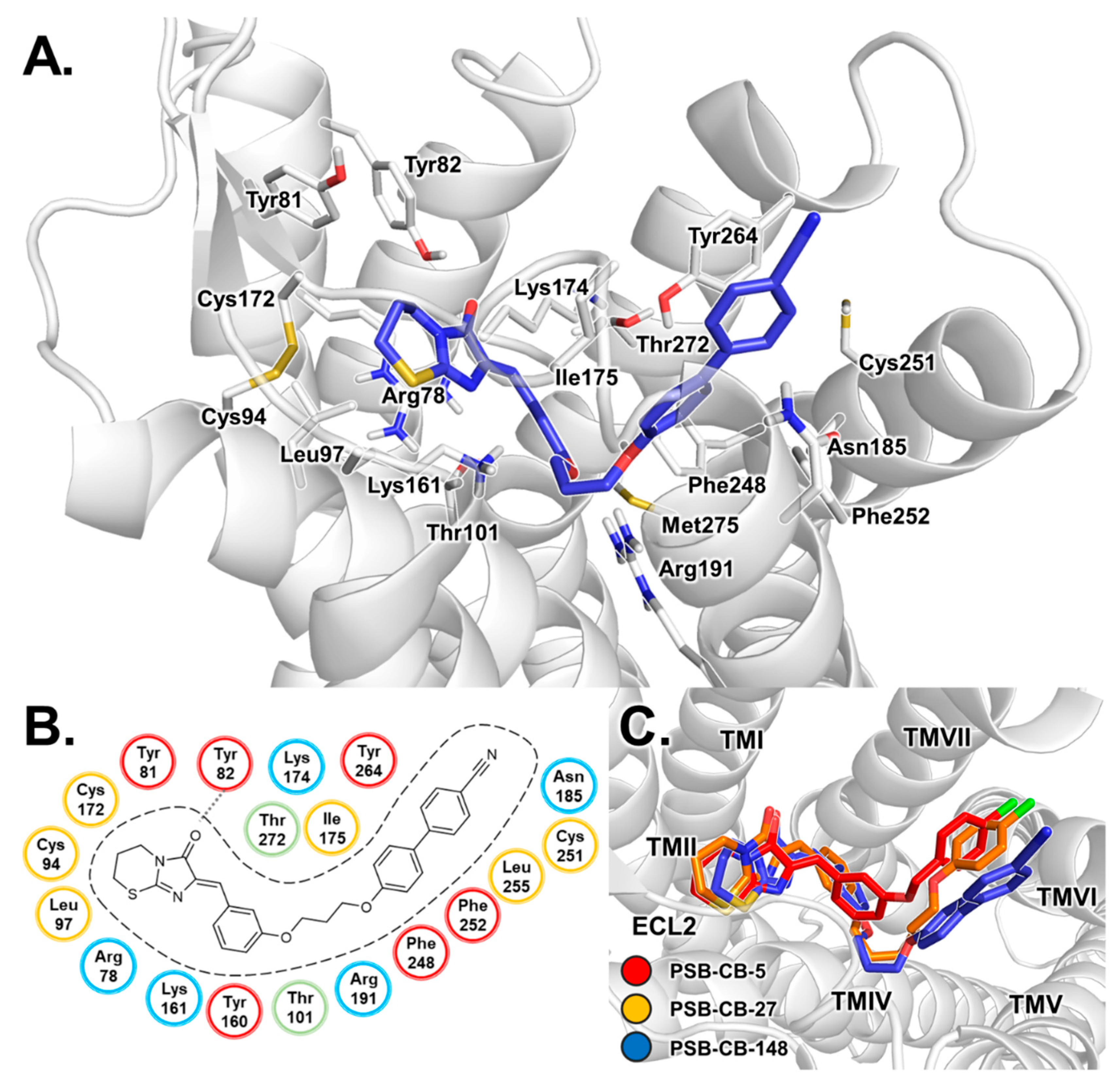
3.3. Binding Mode of THC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta 2007, 1768, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-L.; Wang, Y.; Li, D.-L.; Luo, J.; Liu, M.-Y. Orphan G protein-coupled receptors (GPCRs): Biological functions and potential drug targets. Acta Pharmacol. Sin. 2012, 33, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Muraoka, A.; Yang, Y.K.; Samuelson, L.C.; Zimmerman, E.M.; Cook, H.; Yamada, T. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 1997, 42, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Sumida, H.; Cyster, J.G. G-Protein Coupled Receptor 18 Contributes to Establishment of the CD8 Effector T Cell Compartment. Front. Immunol. 2018, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sumida, H.; Cyster, J.G. GPR18 is required for a normal CD8αα intestinal intraepithelial lymphocyte compartment. J. Exp. Med. 2014, 211, 2351–2359. [Google Scholar] [CrossRef]
- Becker, A.M.; Callahan, D.J.; Richner, J.M.; Choi, J.; DiPersio, J.F.; Diamond, M.S.; Bhattacharya, D. GPR18 controls reconstitution of mouse small intestine intraepithelial lymphocytes following bone marrow transplantation. PLoS ONE 2015, 10, e0133854. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Klesius, P.H. G-protein coupled receptor 18 (GPR18) in channel catfish: Expression analysis and efficacy as immunostimulant against Aeromonas hydrophila infection. Fish Shellfish Immunol. 2013, 35, 1070–1078. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Reyes-Resina, I.; Navarro, G.; Aguinaga, D.; Canela, E.I.; Schoeder, C.T.; Załuski, M.; Kieć-Kononowicz, K.; Saura, C.A.; Müller, C.E.; Franco, R. Molecular and functional interaction between GPR18 and cannabinoid CB2 G-protein-coupled receptors. Relevance in neurodegenerative diseases. Biochem. Pharmacol. 2018, 157, 169–179. [Google Scholar] [CrossRef]
- McHugh, D. GPR18 in microglia: Implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol. 2012, 167, 1575–1582. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef]
- Haugh, O.; Penman, J.; Irving, A.J.; Campbell, V.A. The emerging role of the cannabinoid receptor family in peripheral and neuro-immune interactions. Curr. Drug Targets 2016, 17, 1834–1840. [Google Scholar] [CrossRef]
- Miller, S.; Leishman, E.; Oehler, O.; Daily, L.; Murataeva, N.; Wager-Miller, J.; Bradshaw, H.; Straiker, A. Evidence for a GPR18 role in diurnal regulation of intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6419–6426. [Google Scholar] [CrossRef]
- Caldwell, M.D.; Hu, S.S.-J.; Viswanathan, S.; Bradshaw, H.; Kelly, M.E.M.; Straiker, A. A GPR18-based signalling system regulates IOP in murine eye. Br. J. Pharmacol 2013, 169, 834–843. [Google Scholar] [CrossRef]
- Haskó, J.; Fazakas, C.; Molnár, J.; Nyúl-Tóth, Á.; Herman, H.; Hermenean, A.; Wilhelm, I.; Persidsky, Y.; Krizbai, I.A. CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier. Int. J. Mol. Sci. 2014, 15, 8063–8074. [Google Scholar] [CrossRef]
- Qin, Y.; Verdegaal, E.M.E.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011, 24, 207–218. [Google Scholar] [CrossRef]
- Noreen, N.; Muhammad, F.; Akhtar, B.; Azam, F.; Anwar, M.I. Is cannabidiol a promising substance for new drug development? A review of its potential therapeutic applications. Crit. Rev. Eukaryot. Gene Expr. 2018, 28, 73–86. [Google Scholar] [CrossRef]
- Console-Bram, L.; Brailoiu, E.; Brailoiu, G.C.; Sharir, H.; Abood, M.E. Activation of GPR18 by cannabinoid compounds: A tale of biased agonism. Br. J. Pharmacol. 2014, 171, 3908–3917. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Schoeder, C.T.; Kaleta, M.; Mahardhika, A.B.; Olejarz-Maciej, A.; Łażewska, D.; Kieć-Kononowicz, K.; Müller, C.E. Structure-activity relationships of imidazothiazinones and analogs as antagonists of the cannabinoid-activated orphan G protein-coupled receptor GPR18. Eur. J. Med. Chem. 2018, 155, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Schoeder, C.T.; Hess, C.; Madea, B.; Meiler, J.; Müller, C.E. Pharmacological evaluation of new constituents of “Spice”: Synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicol. 2018, 36, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Hasegawa, H.; Inoue, A.; Muraoka, M.; Miyazaki, T.; Oka, K.; Yasukawa, M. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun. 2006, 347, 827–832. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef]
- van Lu, B.; Puhl, H.L.; Ikeda, S.R. N-Arachidonyl glycine does not activate G protein-coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol. 2013, 83, 267–282. [Google Scholar]
- Rempel, V.; Atzler, K.; Behrenswerth, A.; Karcz, T.; Schoeder, C.; Hinz, S.; Kaleta, M.; Thimm, D.; Kieć-Kononowicz, K.; Müller, C.E. Bicyclic imidazole-4-one derivatives: A new class of antagonists for the orphan G protein-coupled receptors GPR18 and GPR55. Med. Chem. Commun. 2014, 5, 632–649. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, Z.-G.; Zhang, K.; Kiselev, E.; Crane, S.; Wang, J.; Paoletta, S.; Yi, C.; Ma, L.; Zhang, W.; et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 2015, 520, 317–321. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Yerande, S.G.; Namasivayam, V.; Klapschinski, T.A.; Alnouri, M.W.; El-Tayeb, A.; Müller, C.E. Substituted 4-phenylthiazoles: Development of potent and selective A1, A3 and dual A1/A3 adenosine receptor antagonists. Eur. J. Med. Chem. 2020, 186, 111879. [Google Scholar] [CrossRef]
- Ciancetta, A.; O’Connor, R.D.; Paoletta, S.; Jacobson, K.A. Demystifying P2Y1 Receptor Ligand Recognition through Docking and Molecular Dynamics Analyses. J. Chem. Inf. Model. 2017, 57, 3104–3123. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Belousoff, M.J.; Fletcher, M.M.; Zhang, X.; Khoshouei, M.; Deganutti, G.; Koole, C.; Furness, S.G.B.; Miller, L.J.; Hay, D.L.; et al. Structure and Dynamics of Adrenomedullin Receptors AM1 and AM2 Reveal Key Mechanisms in the Control of Receptor Phenotype by Receptor Activity-Modifying Proteins. ACS Pharmacol. Transl. Sci. 2020, 3, 263–284. [Google Scholar] [CrossRef]
- Tosh, D.K.; Rao, H.; Bitant, A.; Salmaso, V.; Mannes, P.; Lieberman, D.I.; Vaughan, K.L.; Mattison, J.A.; Rothwell, A.C.; Auchampach, J.A.; et al. Design and in Vivo Characterization of A1 Adenosine Receptor Agonists in the Native Ribose and Conformationally Constrained (N)-Methanocarba Series. J. Med. Chem. 2019, 62, 1502–1522. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chaudhari, R.; Heim, A.J.; Li, Z. Improving homology modeling of G-protein coupled receptors through multiple-template derived conserved inter-residue interactions. J. Comput. Aided Mol. Des. 2015, 29, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.; Wallner, B.; Lindahl, E.; Elofsson, A. Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 2008, 17, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Kuder, K.J.; Karcz, T.; Kaleta, M.; Kieć-Kononowicz, K. Molecular Modeling of an Orphan GPR18 Receptor. Lett. Drug Des. Discov. 2019, 16, 1167–1174. [Google Scholar] [CrossRef]
- Sotudeh, N.; Morales, P.; Hurst, D.P.; Lynch, D.L.; Reggio, P.H. Towards a molecular understanding of the cannabinoid related orphan receptor GPR18: A focus on its constitutive activity. Int. J. Mol. Sci. 2019, 20, 2300. [Google Scholar] [CrossRef]
- Schmeisser, M.G.; Pearsall, E.A.; Reggio, P.H. Construction of a GPR18 receptor model using conformational memories. Biophys. J. 2013, 104, 409a. [Google Scholar] [CrossRef]
- Reynolds, J.L. Inhibition of GPR18 through docking of known antagonists using a homology model. Biophys. J. 2015, 108, 513a. [Google Scholar] [CrossRef]
- Kothandan, G.; Cho, S.J. Homology modeling of GPR18 Receptor, an orphan G-protein-coupled receptor. J. Chosun Nat. Sci. 2013, 6, 16–20. [Google Scholar] [CrossRef][Green Version]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Gacasan, S.B.; Baker, D.L.; Parrill, A.L. G protein-coupled receptors: The evolution of structural insight. AIMS Biophys. 2017, 4, 491–527. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, Q.; Wu, B. Structural studies of G protein-coupled receptors. Mol. Cells 2015, 38, 836–842. [Google Scholar] [PubMed]
- Grossfield, A. Recent progress in the study of G protein-coupled receptors with molecular dynamics computer simulations. Biochim. Biophys. Acta 2011, 1808, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fu, W.; Wang, Z.; Wang, X.; Lei, T.; Zhu, F.; Li, D.; Chang, S.; Xu, L.; Hou, T. Reliability of docking-based virtual screening for GPCR ligands with homology modeled structures: A case study of the angiotensin II type I receptor. ACS Chem. Neurosci. 2019, 10, 677–689. [Google Scholar] [CrossRef]
- Ciancetta, A.; Rubio, P.; Lieberman, D.I.; Jacobson, K.A. A3 adenosine receptor activation mechanisms: Molecular dynamics analysis of inactive, active, and fully active states. J. Comput. Aided Mol. Des. 2019, 33, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Kashani-Amin, E.; Sakhteman, A.; Larijani, B.; Ebrahim-Habibi, A. Presence of carbohydrate binding modules in extracellular region of class C G-protein coupled receptors (C GPCR): An in silico investigation on sweet taste receptor. J. Biosci. 2019, 44, 138. [Google Scholar] [CrossRef]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How ligands illuminate GPCR molecular pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef]
- Heydenreich, F.M.; Vuckovic, Z.; Matkovic, M.; Veprintsev, D.B. Stabilization of G protein-coupled receptors by point mutations. Front. Pharmacol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Deganutti, G.; Moro, S.; Reynolds, C.A. Peeking at G-protein-coupled receptors through the molecular dynamics keyhole. Future Med. Chem. 2019, 11, 599–615. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Vogel, H.; Filipek, S.; Stevens, R.C.; Palczewski, K. The molecular mechanism of P2Y1 receptor activation. Angew. Chem. 2016, 55, 10331–10335. [Google Scholar] [CrossRef]
- Ribeiro, J.M.L.; Filizola, M. Insights from molecular dynamics simulations of a number of G-Protein coupled receptor targets for the treatment of pain and opioid use disorders. Front. Mol. Neurosci. 2019, 12, 207. [Google Scholar] [CrossRef]
- An, X.; Bai, Q.; Bing, Z.; Zhou, S.; Shi, D.; Liu, H.; Yao, X. How does agonist and antagonist binding lead to different conformational ensemble equilibria of the κ-opioid receptor: Insight from long-time gaussian accelerated molecular dynamics simulation. ACS Chem. Neurosci. 2019, 10, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Tautermann, C.S.; Seeliger, D.; Kriegl, J.M. What can we learn from molecular dynamics simulations for GPCR drug design? Comput. Struct. Biotechnol. J. 2015, 13, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Renault, P.; Louet, M.; Marie, J.; Labesse, G.; Floquet, N. Molecular dynamics simulations of the allosteric modulation of the adenosine A2a receptor by a mini-G Protein. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Molecular mechanisms of allosteric probe dependence in μ opioid receptor. J. Biomol. Struct. Dyn. 2019, 37, 36–47. [Google Scholar] [CrossRef]
- Dalton, J.A.R.; Lans, I.; Giraldo, J. Quantifying conformational changes in GPCRs: Glimpse of a common functional mechanism. BMC Bioinform. 2015, 16, 124. [Google Scholar] [CrossRef]
- Lee, Y.; Basith, S.; Choi, S. Recent advances in structure-based drug design targeting class A G protein-coupled receptors utilizing crystal structures and computational simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef]
- Ballesteros, J.; Kitanovic, S.; Guarnieri, F.; Davies, P.; Fromme, B.J.; Konvicka, K.; Chi, L.; Millar, R.P.; Davidson, J.S.; Weinstein, H.; et al. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J. Biol. Chem. 1998, 273, 10445–10453. [Google Scholar] [CrossRef]
- Wiley, J.L.; Marusich, J.A.; Thomas, B.F. Combination chemistry: Structure-activity relationships of novel psychoactive cannabinoids. Curr. Top. Behav. Neurosci. 2017, 32, 231–248. [Google Scholar]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular targets of the phytocannabinoids-a complex picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- de Petrocellis, L.; Di Marzo, V. Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: Focus on G-protein-coupled receptors and transient receptor potential channels. J. Neuroimmune Pharmacol. 2010, 5, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Lubin, M.L.; Flores, C.M. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J. Neurosci. 2008, 28, 6231–6238. [Google Scholar] [CrossRef] [PubMed]
- de Petrocellis, L.; Vellani, V.; Schiano-Moriello, A.; Marini, P.; Magherini, P.C.; Orlando, P.; Di Marzo, V. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J. Pharmacol. Exp. Ther. 2008, 325, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Cheng, K.; Cui, T.; Godlewski, G.; Rice, K.; Xu, Y.; Zhang, L. Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nat. Chem. Biol. 2011, 7, 296–303. [Google Scholar] [CrossRef]
- Anavi-Goffer, S.; Baillie, G.; Irving, A.J.; Gertsch, J.; Greig, I.R.; Pertwee, R.G.; Ross, R.A. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J. Biol. Chem. 2012, 287, 91–104. [Google Scholar] [CrossRef]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef]
- Picone, R.P.; Khanolkar, A.D.; Xu, W.; Ayotte, L.A.; Thakur, G.A.; Hurst, D.P.; Abood, M.E.; Reggio, P.H.; Fournier, D.J.; Makriyannis, A. (-)-7′-Isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol. Pharmacol. 2005, 68, 1623–1635. [Google Scholar] [CrossRef]
- Pei, Y.; Mercier, R.W.; Anday, J.K.; Thakur, G.A.; Zvonok, A.M.; Hurst, D.; Reggio, P.H.; Janero, D.R.; Makriyannis, A. Ligand-binding architecture of human CB2 cannabinoid receptor: Evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem. Biol. 2008, 15, 1207–1219. [Google Scholar] [CrossRef]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef]
- Shim, J.-Y.; Bertalovitz, A.C.; Kendall, D.A. Identification of essential cannabinoid-binding domains: Structural insights into early dynamic events in receptor activation. J. Biol. Chem. 2011, 286, 33422–33435. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Tao, Q.; Barnett-Norris, J.; Buehner, K.; Hurst, D.P.; Guarnieri, F.; Reggio, P.H.; Nowell Harmon, K.W.; Cabral, G.A.; Abood, M.E. A critical role for a tyrosine residue in the cannabinoid receptors for ligand recognition. Biochem. Pharmacol. 2002, 63, 2121–2136. [Google Scholar] [CrossRef]
- Murphy, J.W.; Kendall, D.A. Integrity of extracellular loop 1 of the human cannabinoid receptor 1 is critical for high-affinity binding of the ligand CP 55,940 but not SR 141716A. Biochem. Pharmacol. 2003, 65, 1623–1631. [Google Scholar] [CrossRef]
- Munk, C.; Mutt, E.; Isberg, V.; Nikolajsen, L.F.; Bibbe, J.M.; Flock, T.; Hanson, M.A.; Stevens, R.C.; Deupi, X.; Gloriam, D.E. An online resource for GPCR structure determination and analysis. Nat. Methods 2019, 16, 151–162. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, A.; Engel, V.; Mahardhika, A.B.; Schoeder, C.T.; Namasivayam, V.; Kieć-Kononowicz, K.; Müller, C.E. Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18. Biomolecules 2020, 10, 686. https://doi.org/10.3390/biom10050686
Neumann A, Engel V, Mahardhika AB, Schoeder CT, Namasivayam V, Kieć-Kononowicz K, Müller CE. Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18. Biomolecules. 2020; 10(5):686. https://doi.org/10.3390/biom10050686
Chicago/Turabian StyleNeumann, Alexander, Viktor Engel, Andhika B. Mahardhika, Clara T. Schoeder, Vigneshwaran Namasivayam, Katarzyna Kieć-Kononowicz, and Christa E. Müller. 2020. "Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18" Biomolecules 10, no. 5: 686. https://doi.org/10.3390/biom10050686
APA StyleNeumann, A., Engel, V., Mahardhika, A. B., Schoeder, C. T., Namasivayam, V., Kieć-Kononowicz, K., & Müller, C. E. (2020). Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18. Biomolecules, 10(5), 686. https://doi.org/10.3390/biom10050686