Functional Implications of Cathelicidin Antimicrobial Protein in Breast Cancer and Tumor-Associated Macrophage Microenvironment

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of TCGA Data
2.2. Clinical Specimen
2.3. Cell Culture and Transfection
2.4. THP-1 Cell Differentiation into Macrophages
2.5. Cell Proliferation Assay
2.6. RNA Labelling and Microarray Analysis
2.7. qRT-PCR
2.8. Aldehyde Dehydrogenase (ALDH) Activity
2.9. Cell Cycle Analysis
2.10. Apoptosis Assay
2.11. Migration Assay
2.12. Invasion Assay
2.13. Immunofluorescence Staining
2.14. Western Blotting
2.15. Development of Stable CAMP Overexpression Cell Lines
2.16. LL-37 Quantification by Enzyme-Linked Immunosorbent Assay (ELISA)
2.17. Transwell Co-Culture Assay
2.18. Immunohistochemistry (IHC) Staining
2.19. Animal Study
2.20. Statistical Analysis
3. Results
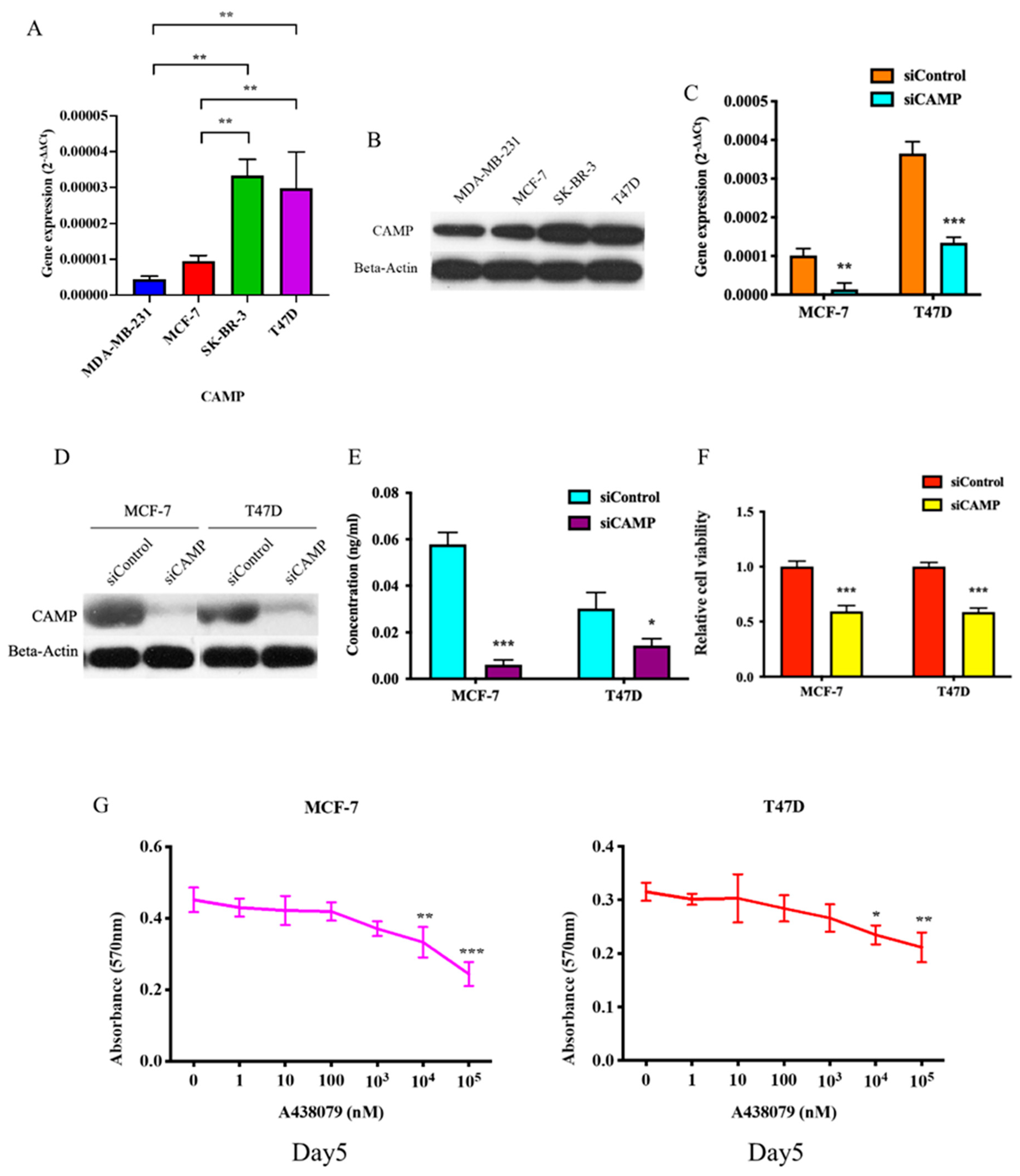
3.1. CAMP mRNA and Cathelicidin Antimicrobial Peptide Are Upregulated in Breast Cancer
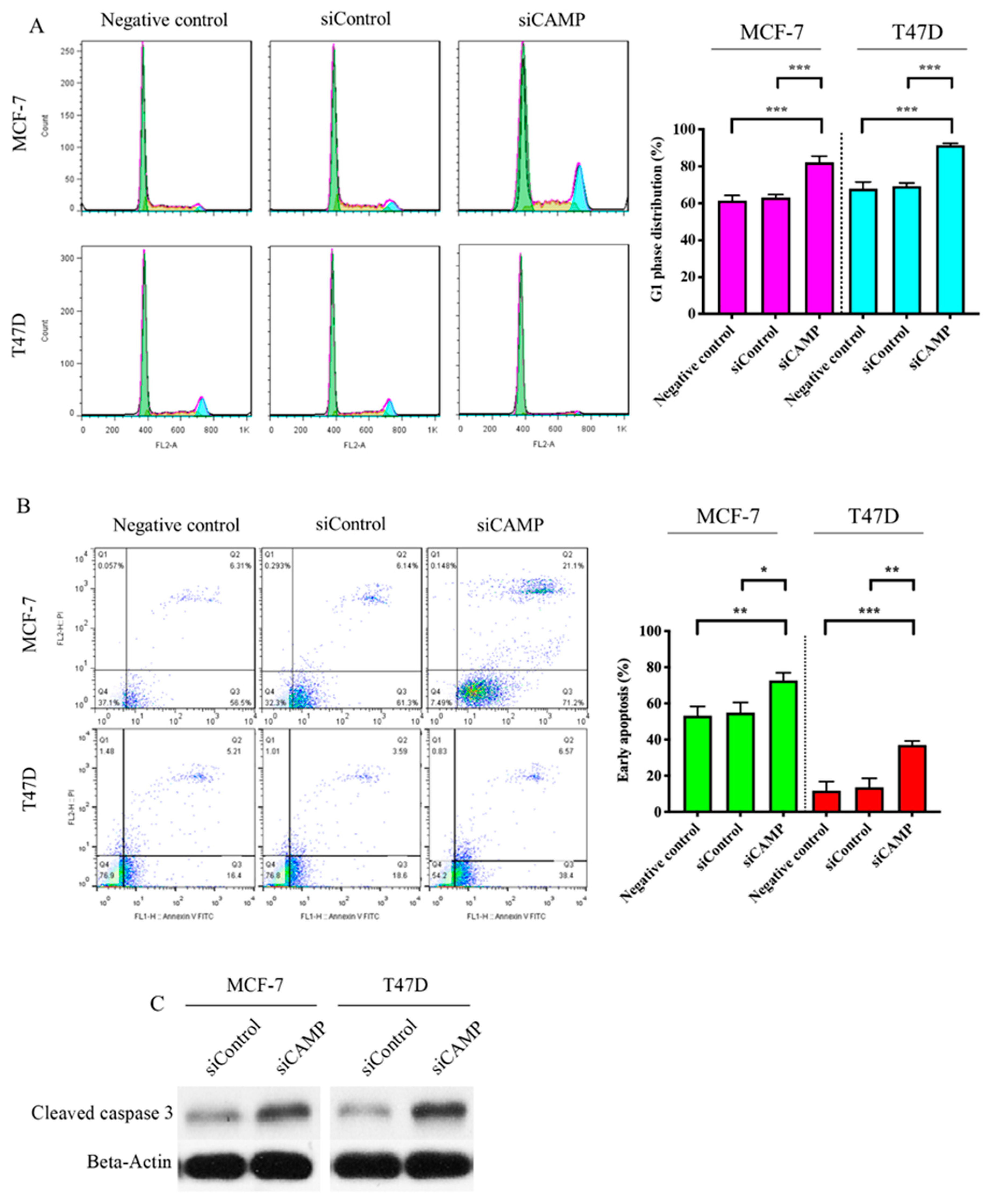
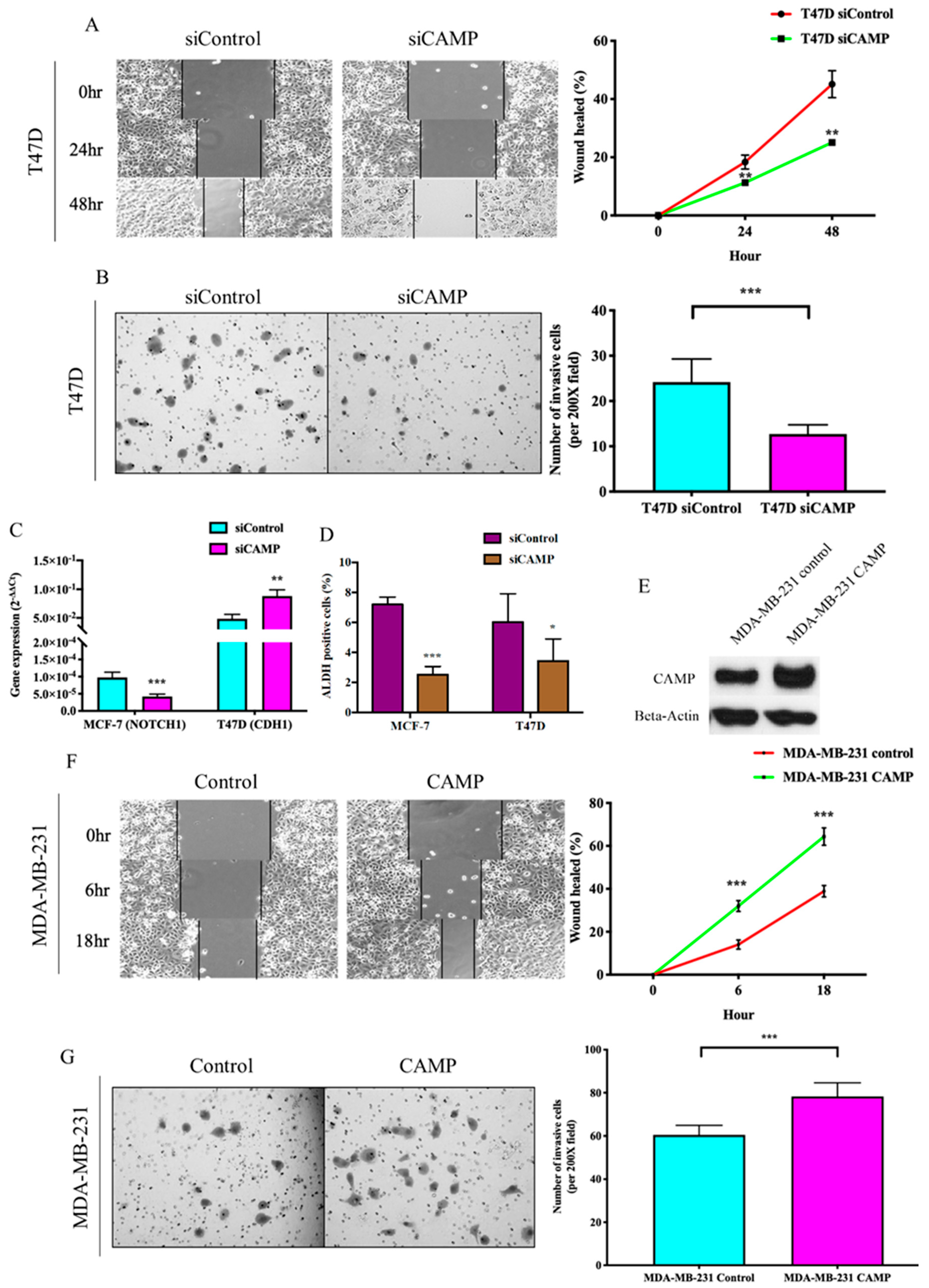
3.2. CAMPConfers Oncogenic Roles in Breast Cancer
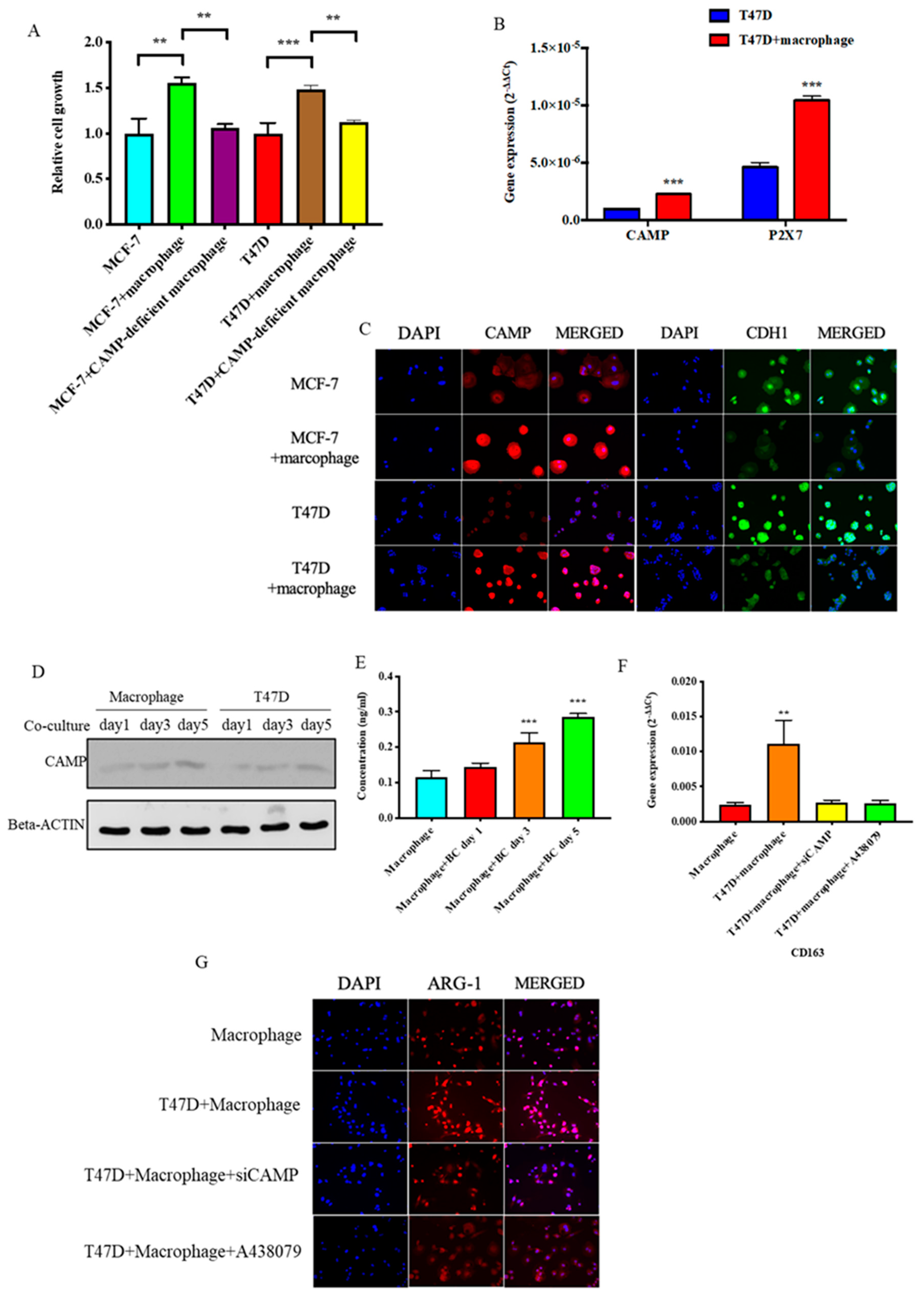
3.3. CAMP is Essential in Tumor Cells-M2 Macrophage Microenvironment
3.4. CAMPInhibition Reduced Tumor Growth In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Globocan. 2018. Available online: http://gco.iarc.fr/today/data/factsheets/cancers/20-Breast-fact-sheet.pdf (accessed on 13 March 2019).
- Wojtowicz, M.E.; Dunn, B.K.; Umar, A. Immunologic approaches to cancer prevention—current status, challenges, and future perspectives. Semin. Oncol. 2016, 43, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.-M.; Weber, B. Genetic and hormonal risk factors in breast cancer. J. Natl. Cancer Inst. 2000, 92, 1126–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begg, L.; Kuller, L.H.; Gutai, J.P.; Caggiula, A.G.; Wolmark, N.; Watson, C.G.; Rao, D.C. Endogenous sex hormone levels and breast cancer risk. Genet. Epidemiol. 1987, 4, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Pike, M.C.; Gerkins, V.R.; Casagrande, J.T.; E Gray, G.; Brown, J.; E Henderson, B. The hormonal basis of breast cancer. Natl. Cancer Inst. Monogr. 1979, 187–193. [Google Scholar]
- Coffelt, S.B.; Waterman, R.S.; Florez, L.; Zu Bentrup, K.H.; Zwezdaryk, K.J.; Tomchuck, S.L.; Lamarca, H.L.; Danka, E.; Morris, C.A.; Scandurro, A.B. Ovarian cancers overexpress the antimicrobial protein hCAP-18 and its derivative LL-37 increases ovarian cancer cell proliferation and invasion. Int. J. Cancer 2007, 122, 1030–1039. [Google Scholar] [CrossRef]
- Agerberth, B.; Gunne, H.; Odeberg, J.; Kogner, P.; Boman, H.G.; Gudmundsson, G.H. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc. Natl. Acad. Sci. USA 1995, 92, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Wang, X.; Zasloff, M.; Wilson, J.M. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. USA 1998, 95, 9541–9546. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, X.; Wu, J.-L.; Quan, W.-Q.; Ma, L.; Yang, F.; Wu, K.-Y.; Wan, H.-Y. Tumor-Produced Versican V1 Enhances hCAP18/LL-37 Expression in Macrophages through Activation of TLR2 and Vitamin D3 Signaling to Promote Ovarian Cancer Progression In Vitro. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Chromek, M.; Slamová, Z.; Bergman, P.; Kovács, L.; Podracká, L.; Ehrén, I.; Hökfelt, T.; Gudmundsson, G.H.; Gallo, R.L.; Agerberth, B.; et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 2006, 12, 636–641. [Google Scholar] [CrossRef]
- Li, D.; Beisswenger, C.; Herr, C.; Schmid, R.M.; Gallo, R.L.; Han, G.; Zakharkina, T.; Bals, R. Expression of the antimicrobial peptide cathelicidin in myeloid cells is required for lung tumor growth. Oncogene 2013, 33, 2709–2716. [Google Scholar] [CrossRef] [Green Version]
- Girnita, A.; Zheng, H.; Grönberg, A.; Girnita, L.; Ståhle, M. Identification of the cathelicidin peptide LL-37 as agonist for the type I insulin-like growth factor receptor. Oncogene 2011, 31, 352–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.X.; Cheng, A.; To, K.F.; Tong, J.H.; Li, M.S.; Shen, J.; Shen, J.; Wong, C.C.; Zhang, L.; Chan, R.L.; et al. Host immune defense peptide LL-37 activates caspase-independent apoptosis and suppresses colon cancer. Cancer Res. 2012, 72, 6512–6523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.X.; Shen, J.; Cheng, A.; Lü, L.; Chan, R.L.Y.; Li, Z.J.; Wang, X.J.; Wong, C.C.M.; Zhang, L.; Ng, S.S.M.; et al. FK-16 Derived from the Anticancer Peptide LL-37 Induces Caspase-Independent Apoptosis and Autophagic Cell Death in Colon Cancer Cells. PLoS ONE 2013, 8, e63641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.E.M.; Hurtado, C.N.L.; Santiago, B.R.; Gonzalez-Amaro, R.; Cañizales, Y.G.C.; Martinez-Fierro, M.L.; Enciso-Moreno, J.A.; Hernández, M.H.G. LL-37, HNP-1, and HBD2/3 modulate the secretion of cytokines TNF-α, IL-6, IFN-γ, IL-10 and MMP1 in human primary cell cultures. Eur. Cytokine Netw. 2016, 27, 68–74. [Google Scholar]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.; Lamarca, H.L.; Tomchuck, S.L.; Zu Bentrup, K.H.; Danka, E.; Henkle, S.L.; et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar] [CrossRef] [Green Version]
- Verjans, E.-T.; Zels, S.; Luyten, W.; Landuyt, B.; Schoofs, L. Molecular mechanisms of LL-37-induced receptor activation: An overview. Peptides 2016, 85, 16–26. [Google Scholar] [CrossRef]
- Bucki, R.; Leszczyńska, K.; Namiot, A.; Sokołowski, W. Cathelicidin LL-37: A Multitask Antimicrobial Peptide. Arch. Immunol. Ther. Exp. 2010, 58, 15–25. [Google Scholar] [CrossRef]
- Hensel, J.A.; Chanda, D.; Kumar, S.; Sawant, A.; Grizzle, W.E.; Siegal, G.P.; Ponnazhagan, S. LL-37 as a therapeutic target for late stage prostate cancer. Prostate 2010, 71, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Craske, M.; Severino, P.; Lima, T.; Labhart, P.; Chammas, R.; Velasco, I.T.; Machado, M.C.C.; Egan, B.; Nakaya, H.I.; et al. Antimicrobial peptide LL-37 participates in the transcriptional regulation of melanoma cells. J. Cancer 2016, 7, 2341–2345. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jia, J.; Li, C.; Duan, Q.; Yang, J.; Wang, X.; Li, R.; Chen, C.; Yan, H.; Zheng, Y. Antimicrobial peptide LL-37 promotes the proliferation and invasion of skin squamous cell carcinoma by upregulating DNA-binding protein A. Oncol. Lett. 2016, 12, 1745–1752. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Condeelis, J.S. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genin, M.; Clément, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.; Edwards, J. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2008, 9, 239–252. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Mahmoud, S.; Lee, A.H.S.; Paish, E.C.; Macmillan, R.D.; Ellis, I.; Green, A.R. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J. Clin. Pathol. 2011, 65, 159–163. [Google Scholar] [CrossRef]
- Elliott, L.A.; Doherty, G.; Sheahan, K.; Ryan, E. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front. Immunol. 2017, 8, 526. [Google Scholar] [CrossRef]
- Cui, R.; Yue, W.; Lattime, E.C.; Stein, M.N.; Xu, Q.; Tan, X.-L. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 2016, 7, 50735–50754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, H.-R.; Lee, J.H.; Hensel, J.A.; Sawant, A.B.; Davis, B.H.; Lee, C.M.; Deshane, J.S.; Ponnazhagan, S. Prostate cancer-derived cathelicidin-related antimicrobial peptide facilitates macrophage differentiation and polarization of immature myeloid progenitors to protumorigenic macrophages. Prostate 2016, 76, 624–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, H.; Büttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D–dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra47. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Agerberth, B.; Charo, J.; Werr, J.; Olsson, B.; Idali, F.; Lindbom, L.; Kiessling, R.; Jörnvall, H.; Wigzell, H.; Gudmundsson, G. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood 2000, 96, 3086–3093. [Google Scholar] [CrossRef]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell. Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Reinholz, M.; Ruzicka, T.; Schauber, J. Cathelicidin LL-37: An Antimicrobial Peptide with a Role in Inflammatory Skin Disease. Ann. Dermatol. 2012, 24, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Schauber, J.; Coda, A.; Lin, H.; Dorschner, R.; Schechter, N.M.; Bonnart, C.; Descargues, P.; Hovnanian, A.; Gallo, R.L. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 2006, 20, 2068–2080. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, D.; Rayner, J.; Couto, M.; Ganz, T. Cationic defensins arise from charge-neutralized propeptides: A mechanism for avoiding leukocyte autocytotoxicity? J. Leukoc. Boil. 1992, 51, 634–639. [Google Scholar] [CrossRef]
- Schauber, J.; Gallo, R.L. Expanding the roles of antimicrobial peptides in skin: Alarming and arming keratinocytes. J. Investig. Dermatol. 2007, 127, 510–512. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, O.; Arnljots, K.; Cowland, J.B.; Bainton, D.F.; Borregaard, N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 1997, 90, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Büchau, A.S.; Morizane, S.; Trowbridge, J.; Schauber, J.; Kotol, P.; Bui, J.D.; Gallo, R.L. The host defense peptide cathelicidin is required for NK cell-mediated suppression of tumor growth. J. Immunol. 2009, 184, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Vitiello, A.; Gallo, R.L. Cutting edge: Mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 2003, 170, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.; Santos, J.C.; Mishra, B.B.; Jena, P.; Progida, C.; Sorensen, O.E.; Gallo, R.L.; Appelberg, R.; Griffiths, G. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell. Microbiol. 2011, 13, 1601–1617. [Google Scholar] [CrossRef] [PubMed]
- Kusaka, S.; Nishida, A.; Takahashi, K.; Bamba, S.; Yasui, H.; Kawahara, M.; Inatomi, O.; Sugimoto, M.; Andoh, A. Expression of human cathelicidin peptide LL-37 in inflammatory bowel disease. Clin. Exp. Immunol. 2017, 191, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Frasca, L.; Palazzo, R.; Chimenti, M.S.; Alivernini, S.; Tolusso, B.; Bui, L.; Botti, E.; Giunta, A.; Bianchi, L.; Petricca, L.; et al. Anti-LL37 Antibodies Are Present in Psoriatic Arthritis (PsA) Patients: New Biomarkers in PsA. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Sahebari, M.; Roshandel, G.; Saadati, N.; Saghafi, M.; Abdolahi, N.; Rezaieyazdi, Z. Cathelicidin (LL-37) and its correlation with pro-oxidant, antioxidant balance and disease activity in systemic lupus erythematosus: A cross-sectional human study. Lupus 2017, 26, 975–982. [Google Scholar] [CrossRef]
- Edfeldt, K.; Agerberth, B.; Rottenberg, M.; Gudmundsson, G.H.; Wang, X.-B.; Mandal, K.; Xu, Q.; Yan, Z.-Q. Involvement of the Antimicrobial Peptide LL-37 in Human Atherosclerosis. Arter. Thromb. Vasc. Boil. 2006, 26, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Shaykhiev, R.; Beißwenger, C.; Kändler, K.; Senske, J.; Püchner, A.; Damm, T.; Behr, J.; Bals, R. Human endogenous antibiotic LL-37 stimulates airway epithelial cell proliferation and wound closure. Am. J. Physiol. Cell. Mol. Physiol. 2005, 289, L842–L848. [Google Scholar] [CrossRef]
- Heilborn, J.D.; Nilsson, M.F.; Jimenez, C.I.C.; Sandstedt, B.; Borregaard, N.; Tham, E.; Sorensen, O.E.; Weber, G.; Ståhle, M. Antimicrobial protein hCAP18/LL-37 is highly expressed in breast cancer and is a putative growth factor for epithelial cells. Int. J. Cancer 2004, 114, 713–719. [Google Scholar] [CrossRef]
- Chen, X.; Zou, X.; Qi, G.; Tang, Y.; Guo, Y.; Si, J.; Liang, L. Roles and Mechanisms of Human Cathelicidin LL-37 in Cancer. Cell. Physiol. Biochem. 2018, 47, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Carmona, F.J.; Montemurro, F.; Kannan, S.; Rossi, V.; Verma, C.; Baselga, J.; Scaltriti, M. AKT signaling in ERBB2-amplified breast cancer. Pharmacol. Ther. 2015, 158, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farabaugh, S.M.; Chan, B.T.; Cui, X.; Dearth, R.K.; Lee, A.V. Lack of interaction between ErbB2 and insulin receptor substrate signaling in breast cancer. Cell Commun. Signal. 2016, 14, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worden, F.P.; Sacco, A.G. Molecularly targeted therapy for the treatment of head and neck cancer: A review of the ErbB family inhibitors. OncoTargets Ther. 2016, 9, 1927–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, G.; Chamorro, C.I.; Granath, F.; Liljegren, A.; Zreika, S.; Saidak, Z.; Sandstedt, B.; Rotstein, S.; Mentaverri, R.; Sánchez, F.; et al. Human antimicrobial protein hCAP18/LL-37 promotes a metastatic phenotype in breast cancer. Breast Cancer Res. 2009, 11, R6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainz, B.; Alcala, S.; Garcia, E.; Sanchez-Ripoll, Y.; Azevedo, M.; Cioffi, M.; Tatari, M.; Miranda-Lorenzo, I.; Hidalgo, M.; López, G.G.; et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015, 64, 1921–1935. [Google Scholar] [CrossRef] [Green Version]
- Neto, G.C.; De Lima, T.M.; Barbeiro, H.V.; Chammas, R.; Machado, M.C.C.; Da Silva, F.P. Cathelicidin LL-37 Promotes or Inhibits Cancer Cell Stemness Depending on the Tumor Origin. Oncomedicine 2016, 1, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics 2014, 4, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Poon, G.F.T.; Birkenhead, D.; Peña, O.M.; Falsafi, R.; Dahlgren, C.; Karlsson, A.; Bylund, J.; Hancock, R.E.W.; Johnson, P. Host Defense Peptide LL-37 Selectively Reduces Proinflammatory Macrophage Responses. J. Immunol. 2011, 186, 5497–5505. [Google Scholar] [CrossRef] [Green Version]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2012, 31, 760–768. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark. Res. 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, N.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seya, T.; Shime, H.; Matsumoto, M. TAMable tumor-associated macrophages in response to innate RNA sensing. OncoImmunology 2012, 1, 1000–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer (n = 110) | |
|---|---|
| Age (years; mean (SD)) | 49.5 (12.8) |
| Histological type | |
| DCIS | 38 |
| IDC | 72 |
| Bilateral cancer | 6 |
| Stage | |
| 0 | 41 |
| I | 34 |
| II | 25 |
| III | 6 |
| IV | 4 |
| Histological grade | |
| 1 | 8 |
| 2 | 34 |
| 3 | 21 |
| NA | 47 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Shin, V.Y.; Ho, J.C.-W.; Siu, M.-T.; Cheuk, I.W.-Y.; Kwong, A. Functional Implications of Cathelicidin Antimicrobial Protein in Breast Cancer and Tumor-Associated Macrophage Microenvironment. Biomolecules 2020, 10, 688. https://doi.org/10.3390/biom10050688
Chen J, Shin VY, Ho JC-W, Siu M-T, Cheuk IW-Y, Kwong A. Functional Implications of Cathelicidin Antimicrobial Protein in Breast Cancer and Tumor-Associated Macrophage Microenvironment. Biomolecules. 2020; 10(5):688. https://doi.org/10.3390/biom10050688
Chicago/Turabian StyleChen, Jiawei, Vivian Yvonne Shin, John Chi-Wang Ho, Man-Ting Siu, Isabella Wai-Yin Cheuk, and Ava Kwong. 2020. "Functional Implications of Cathelicidin Antimicrobial Protein in Breast Cancer and Tumor-Associated Macrophage Microenvironment" Biomolecules 10, no. 5: 688. https://doi.org/10.3390/biom10050688