D-Loop Mutation G42A/G46A Decreases Actin Dynamics

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
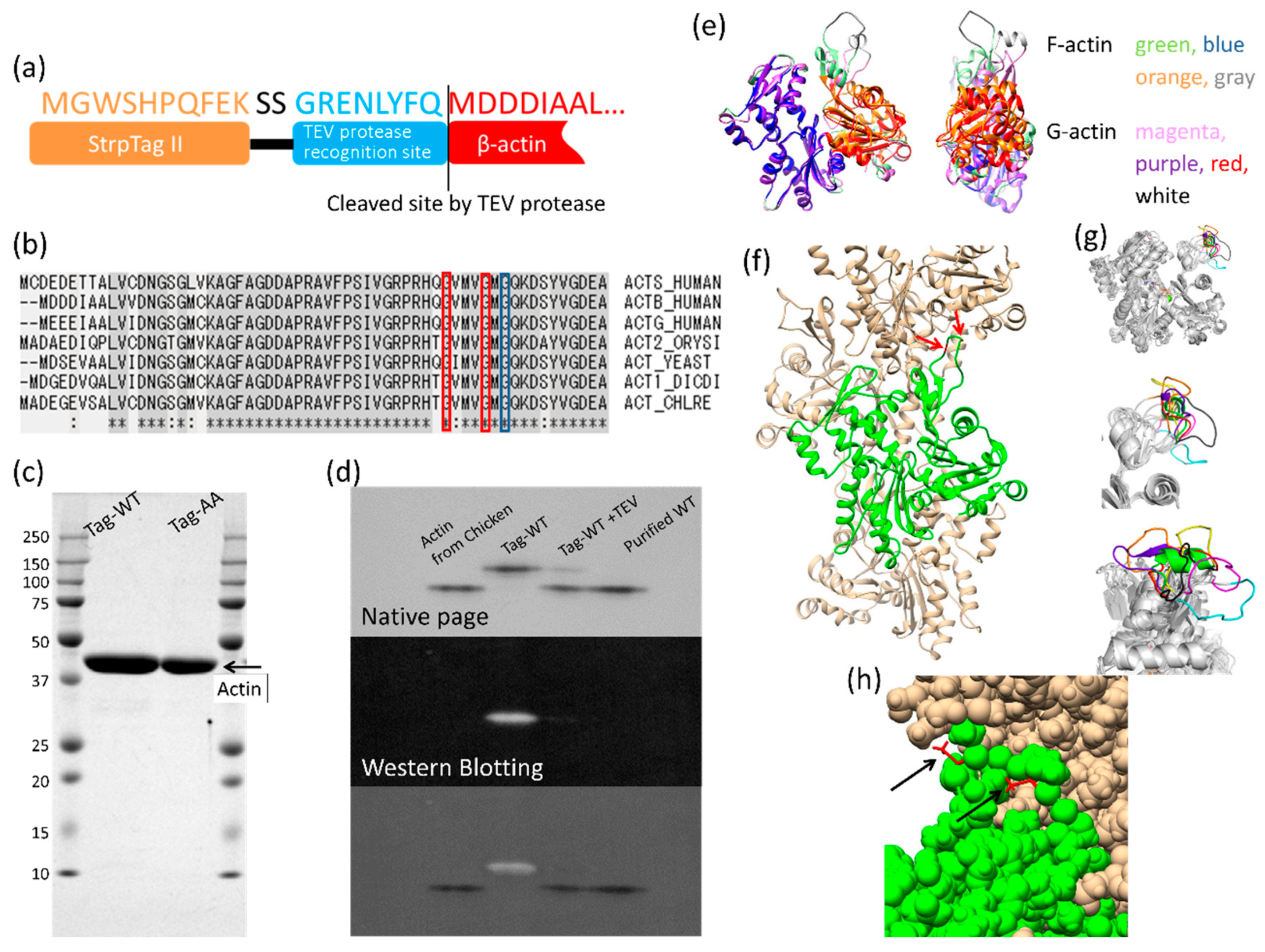
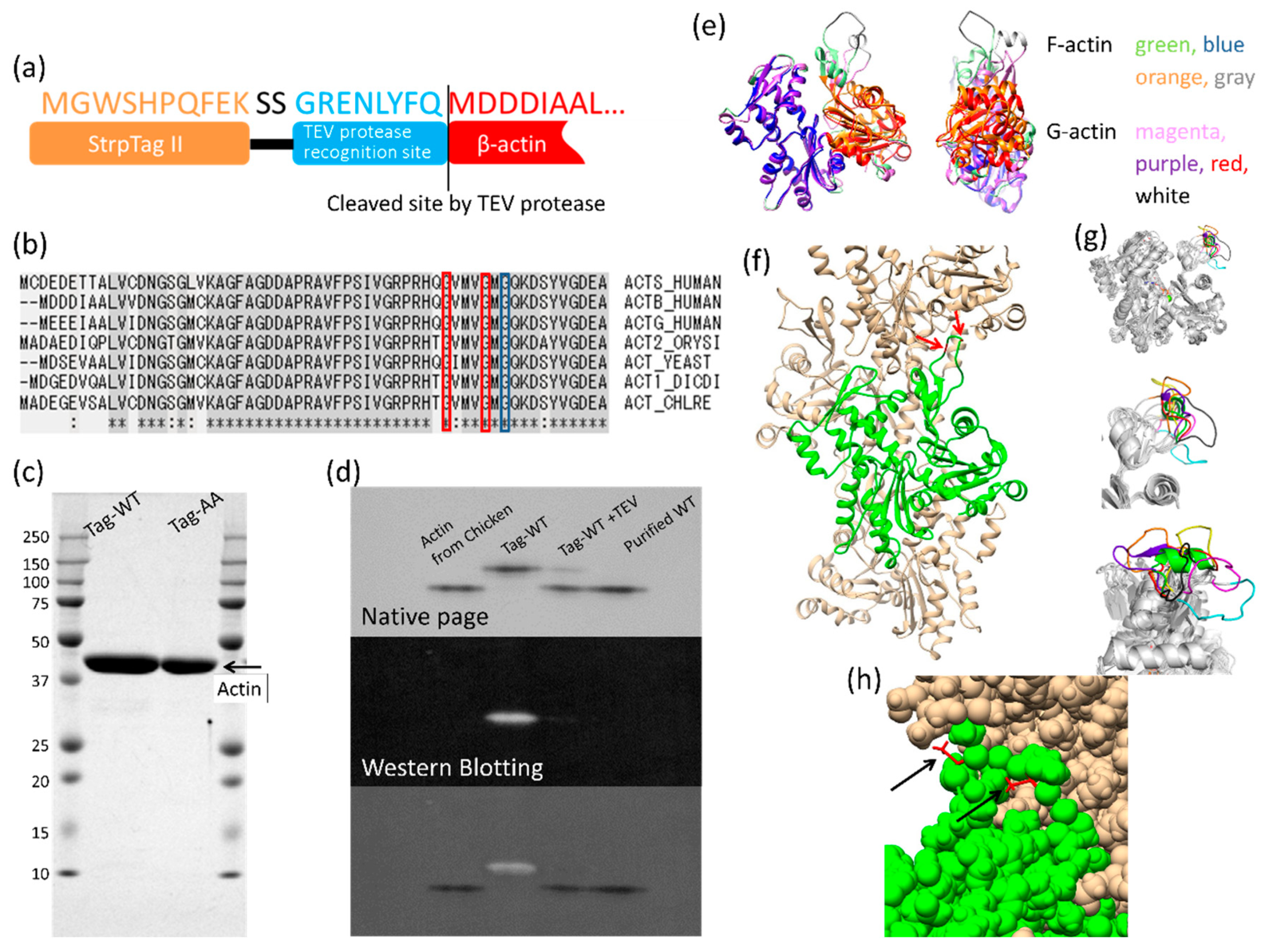
2.1. Construction of the β-Actin Expression Vector
2.2. Expression of Recombinant Human β-Actin in Insect Cells
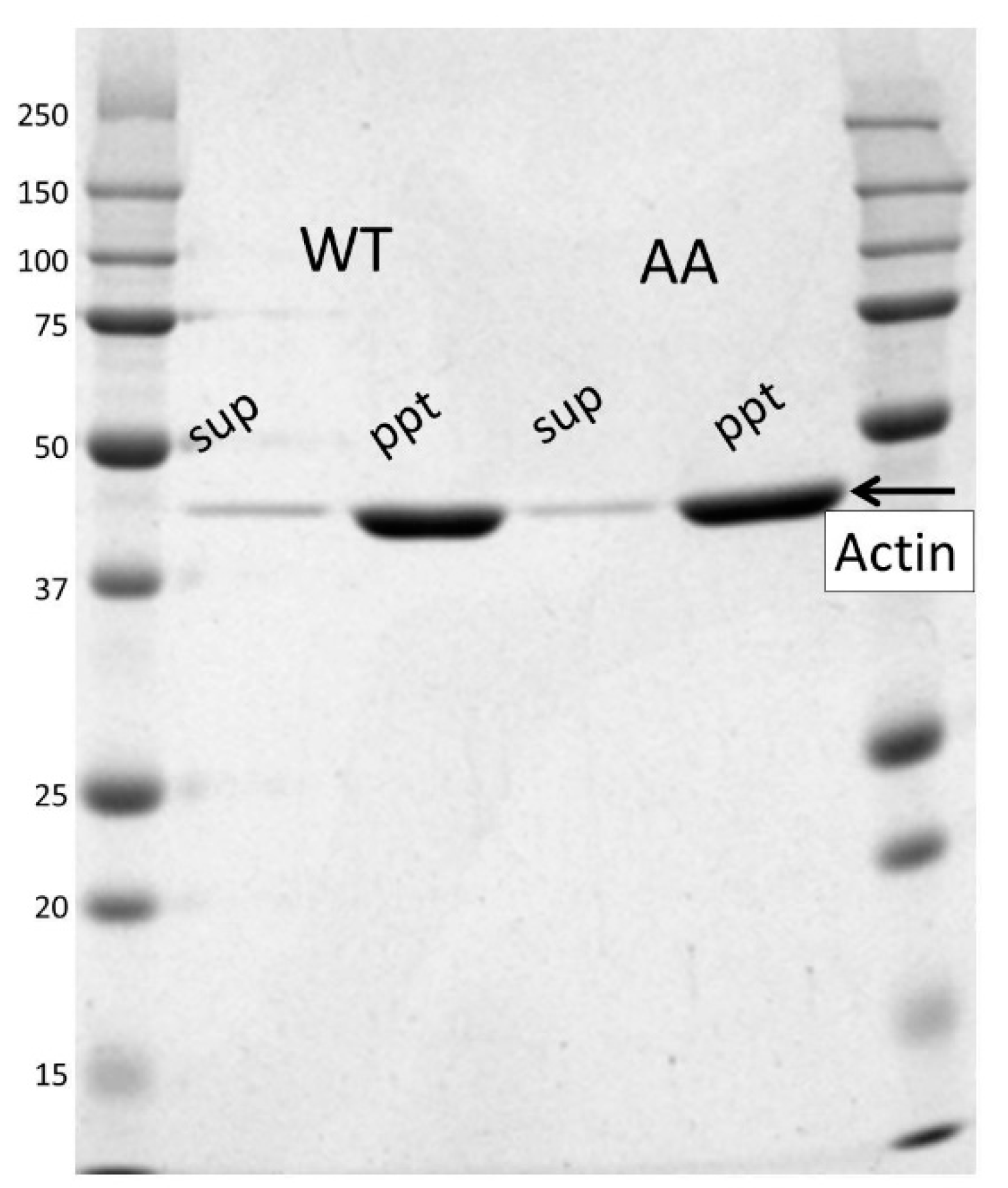
2.3. Purification of Recombinant Actins
2.4. Native-PAGE
2.5. Western Blotting
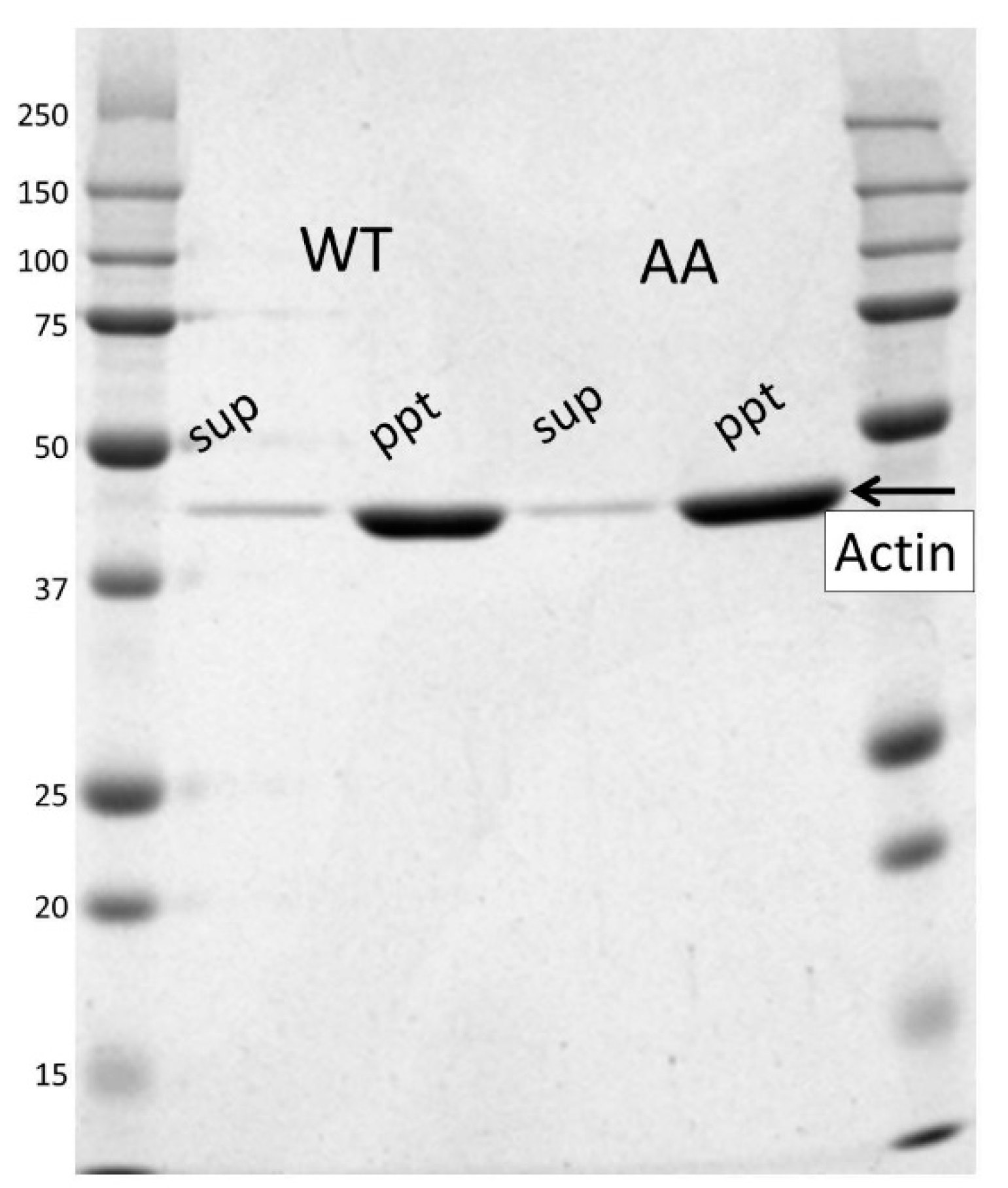
2.6. Critical Concentrations
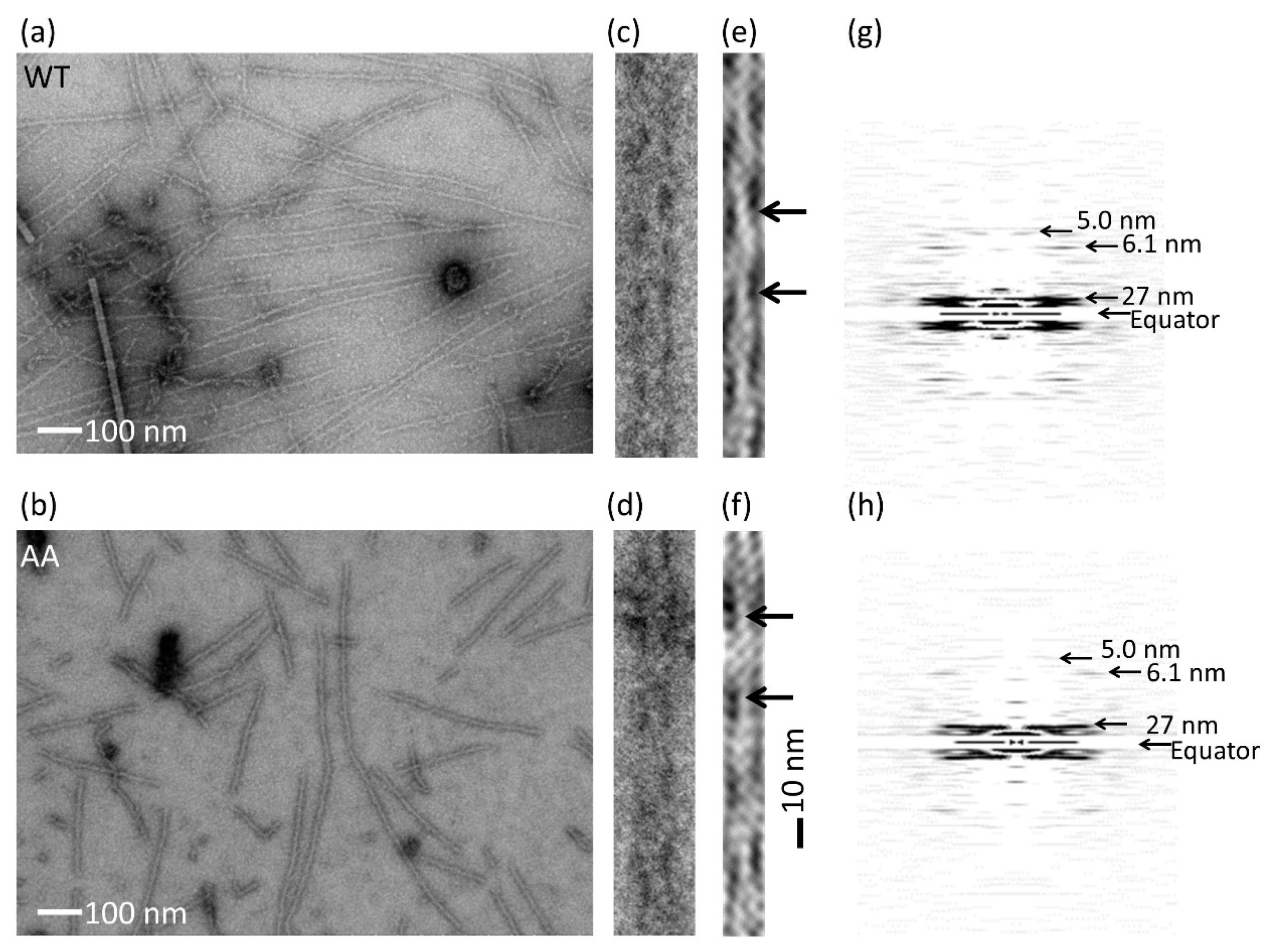
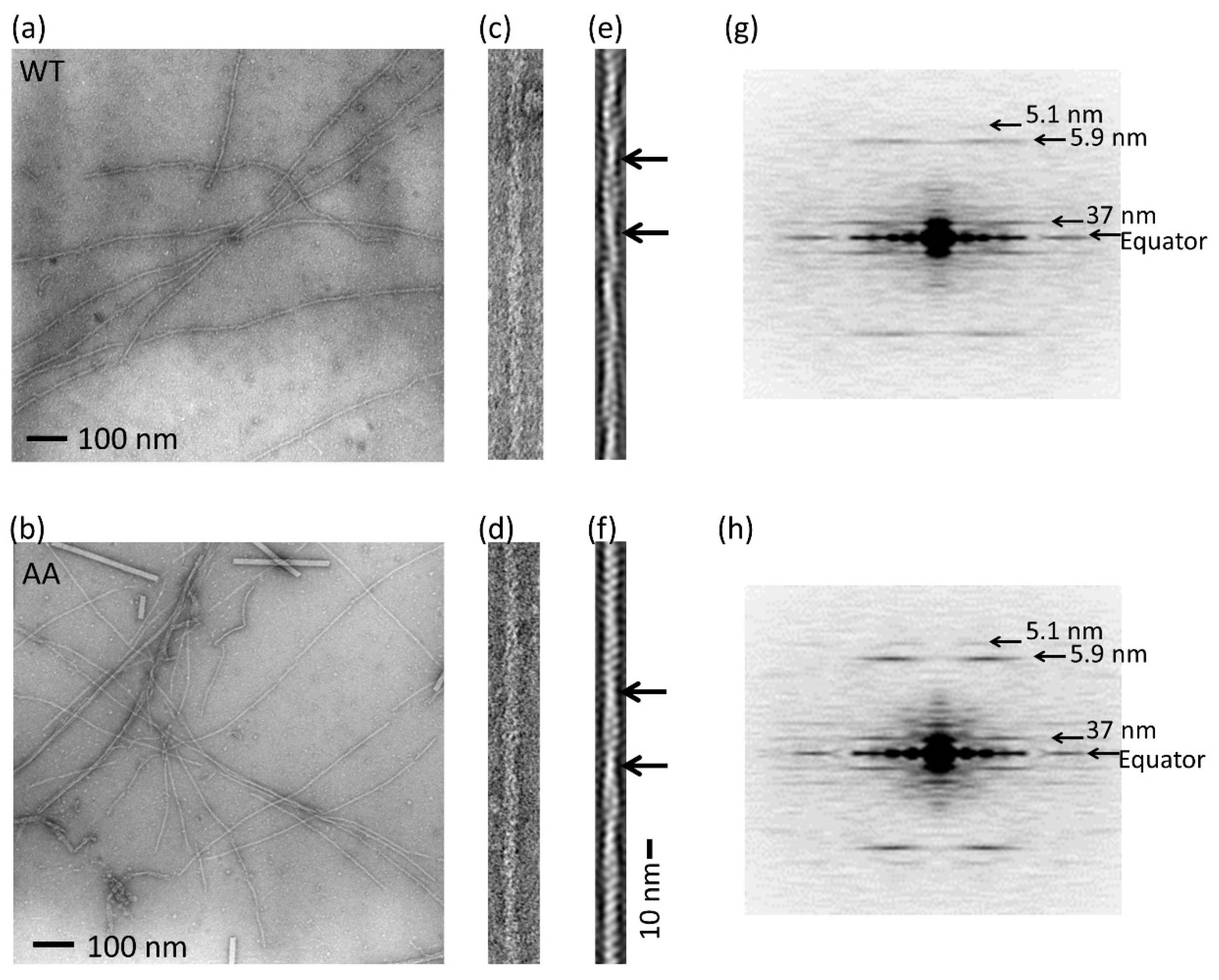
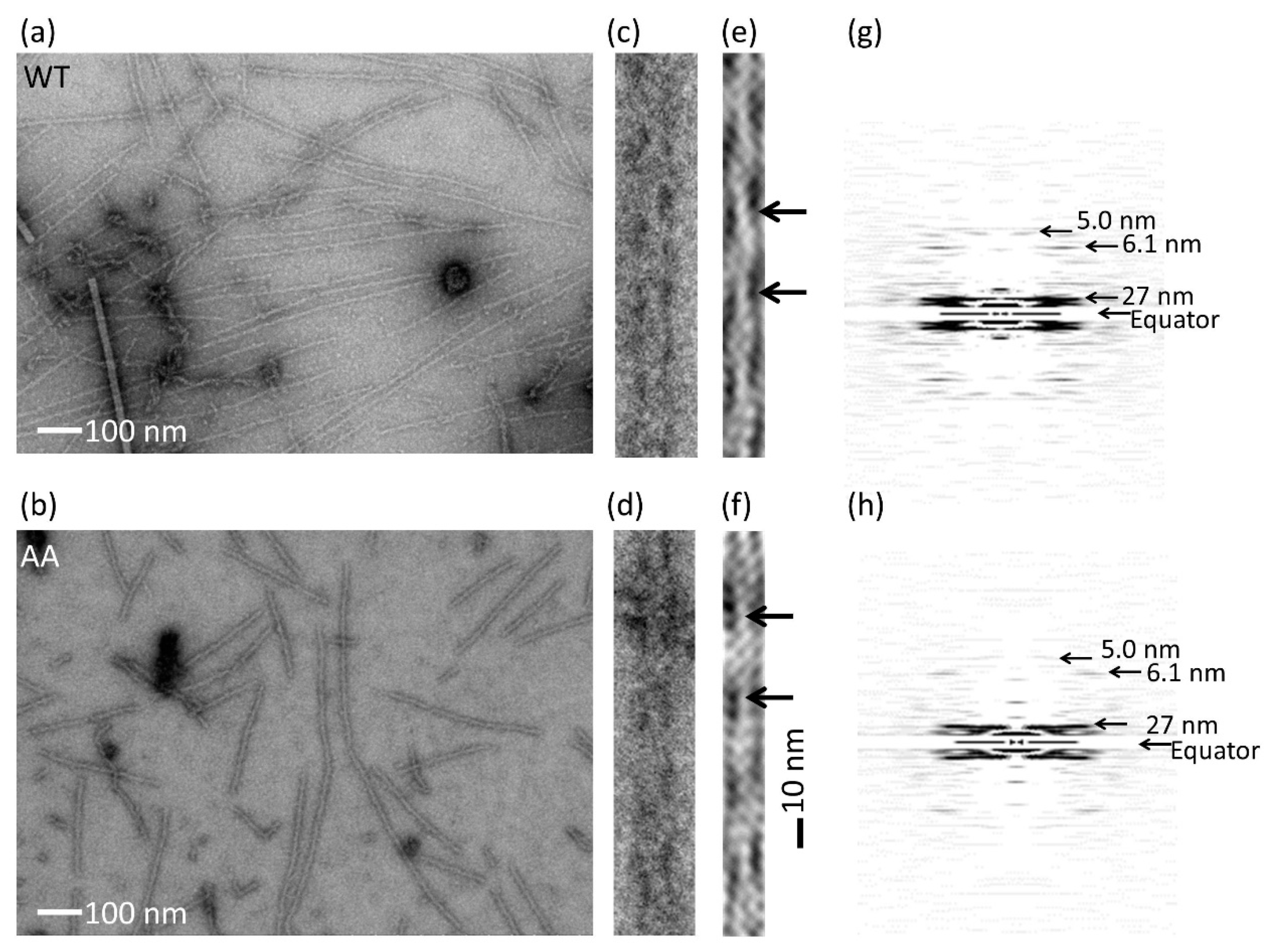
2.7. Electron Microscopy
2.8. Proteins and Protein Labeling for Total Internal Reflection Fluorescence (TIRF) Microscopy
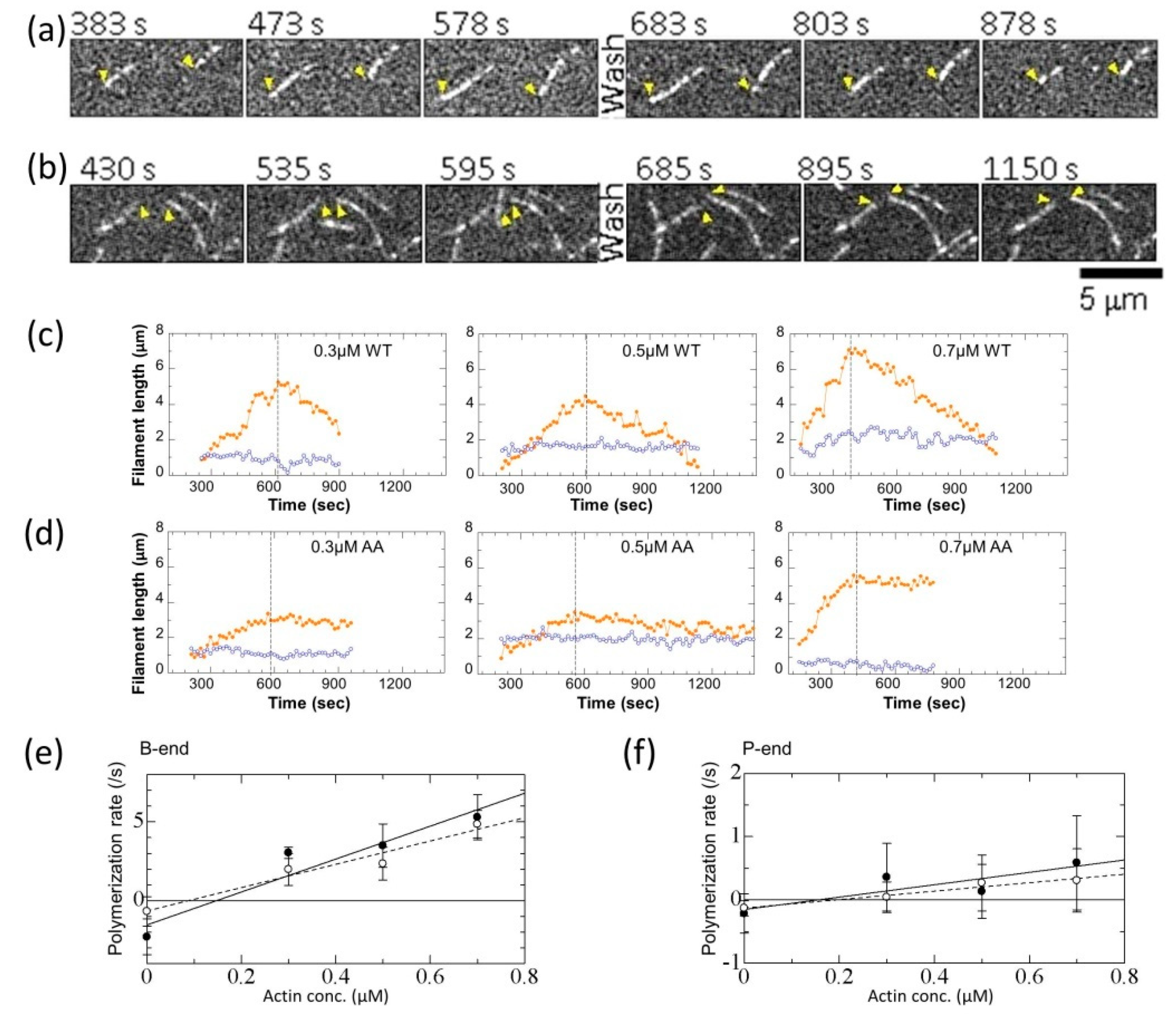
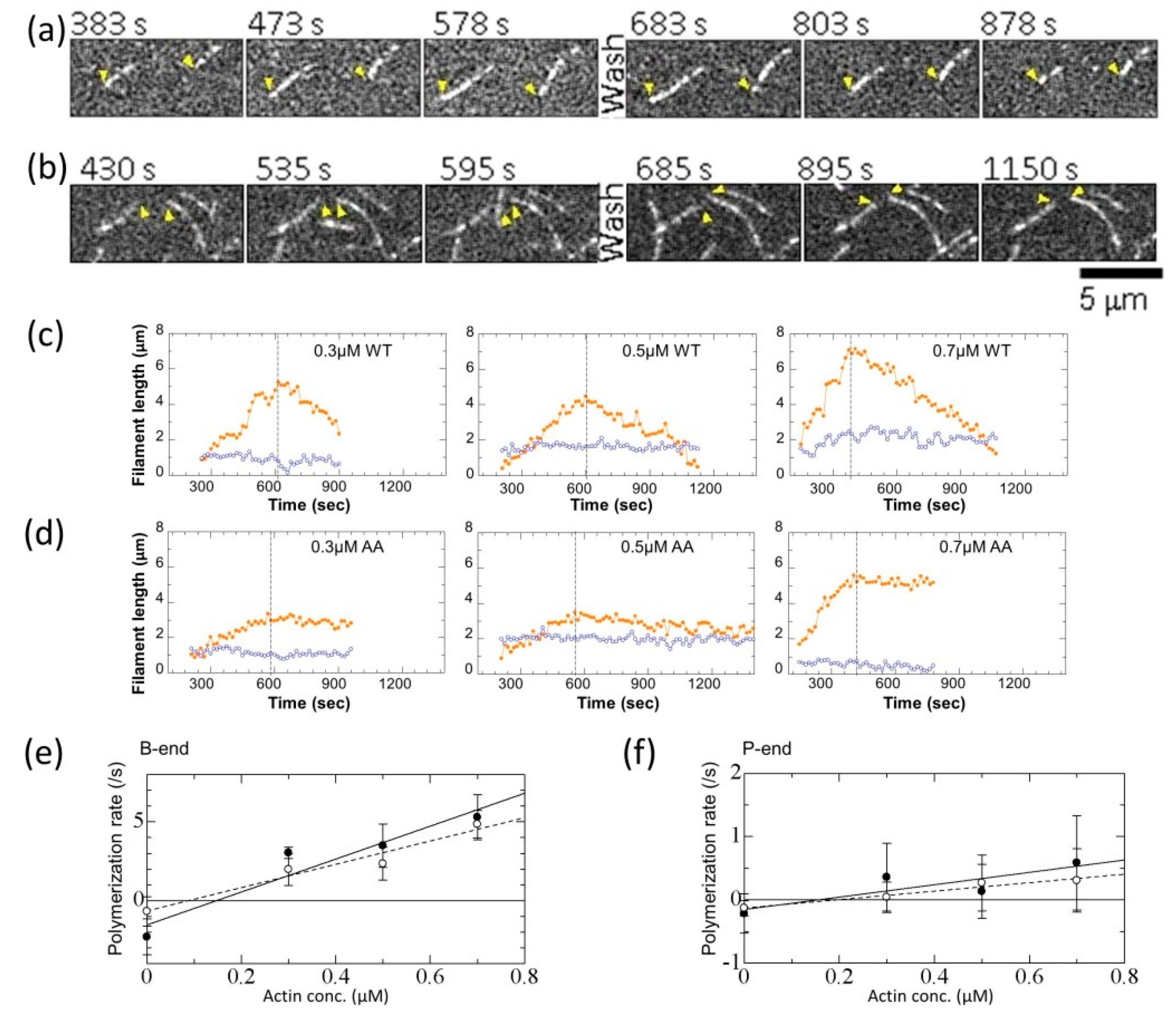
2.9. TIRFAssay and Image Analysis
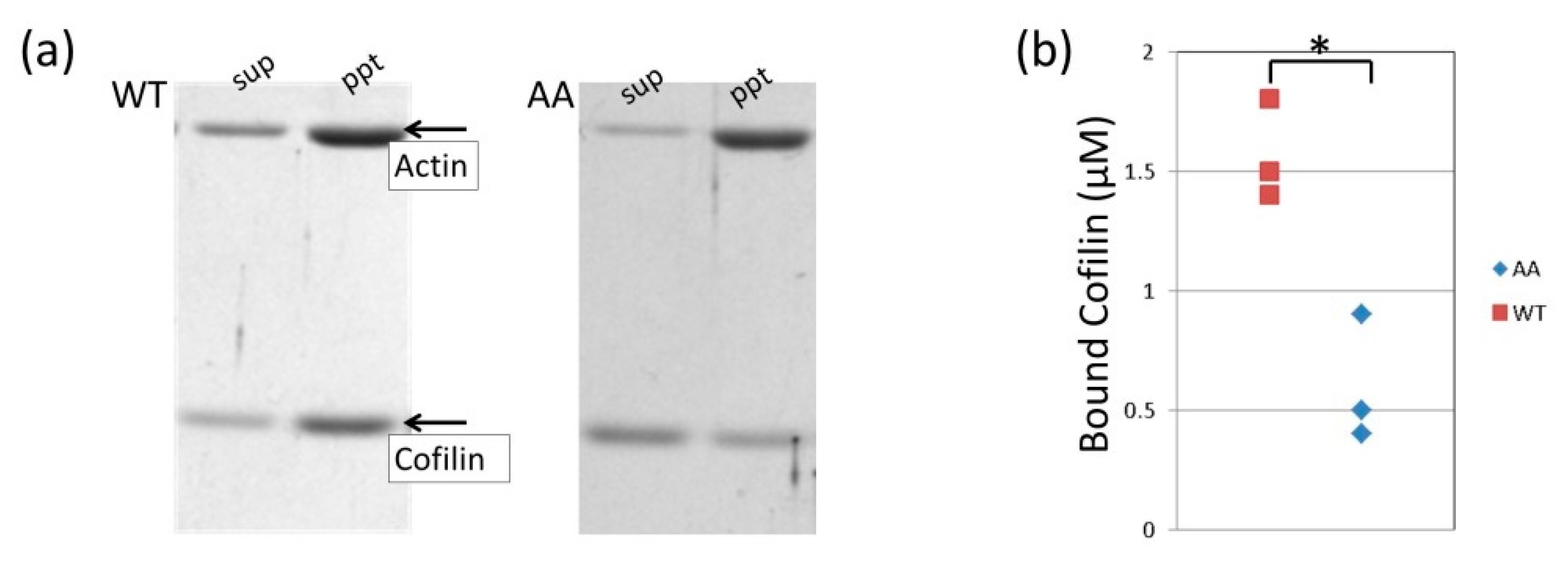
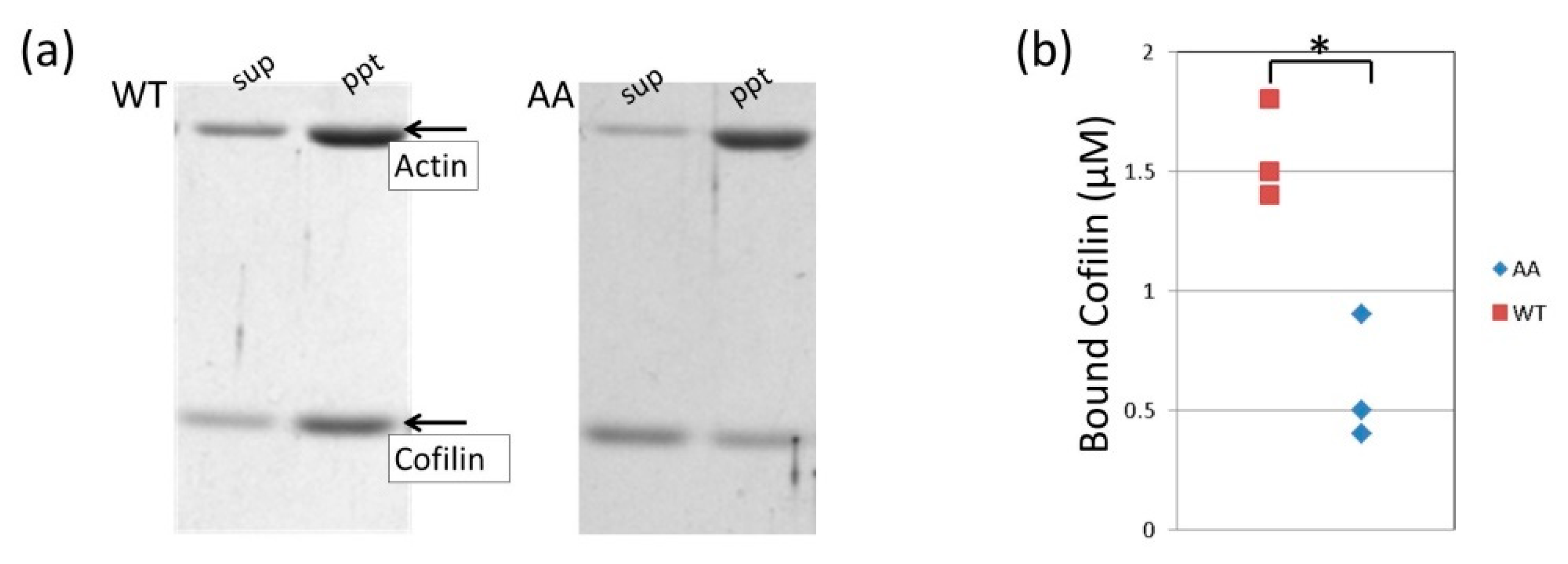
2.10. Co-Sedimentation Assay
2.11. Image Analysis
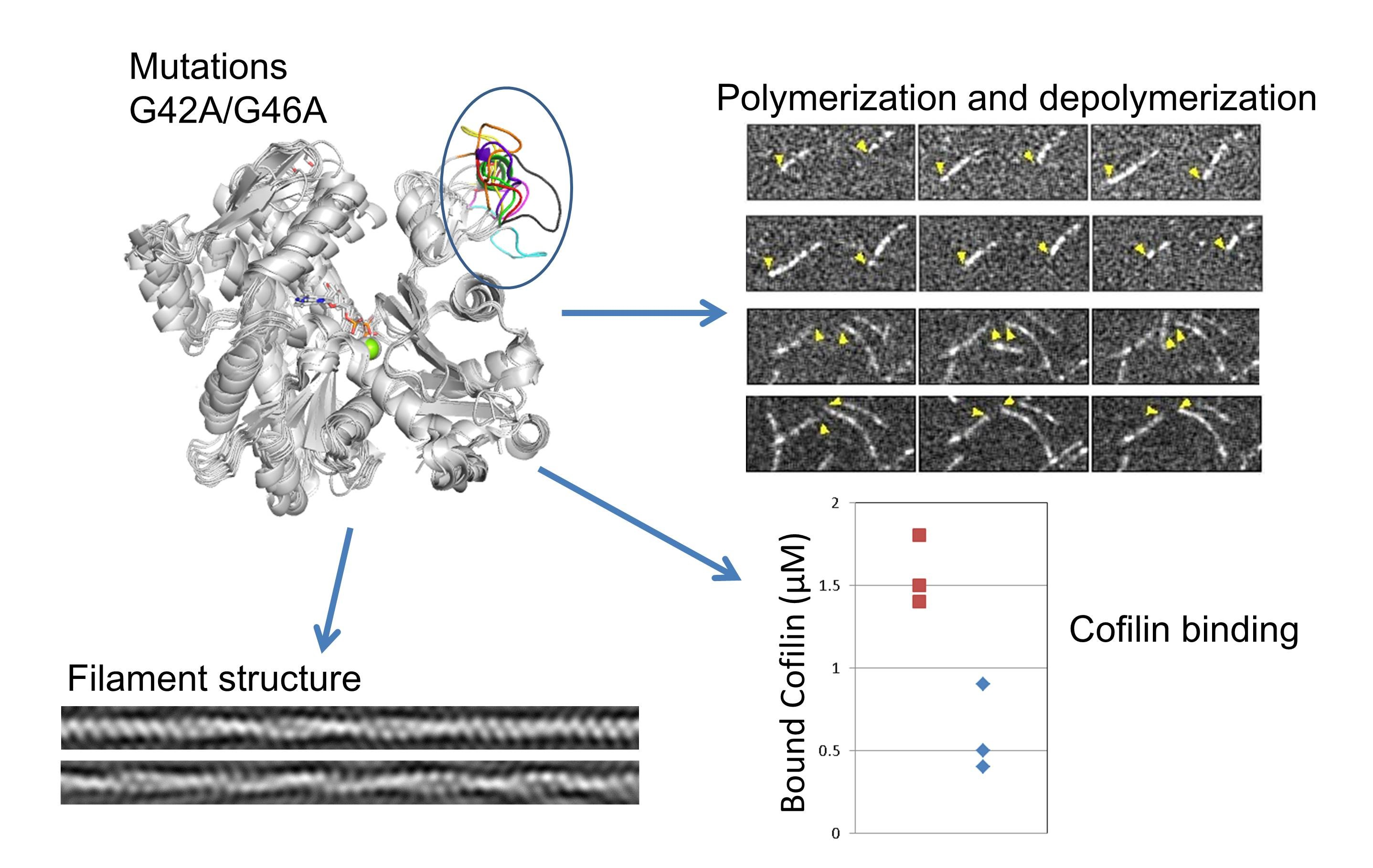
3. Results
3.1. Expression and Purification of Recombinant Human Cytoskeletal β-Actins in Insect Cells
3.2. Effects of the G42A/G46A Mutations on Subunit–Subunit Interactions
3.3. Effects of the G42A/G46A Mutations on Polymerization and Depolymerization Rates
3.4. Effects of the G42A/G46A Mutations on Cofilin Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheterline, P.; Clayton, J.; Sparrow, J. Actin. Protein Profile 1995, 2, 1–103. [Google Scholar]
- Oda, T.; Takeda, S.; Narita, A.; Maeda, Y. Structural polymorphism of actin. J. Mol. Biol. 2019, 431, 3217–3228. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Takeda, S.; Mitsuoka, K.; Oda, T.; Kimura-Sakiyama, C.; Maeda, Y.; Narita, A. Structural basis for cofilin binding and actin filament disassembly. Nat. Commun. 2018, 9, 1860. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Mannherz, H.G.; Suck, D.; Pai, E.F.; Holmes, K.C. Atomic structure of the actin:Dnase i complex. Nature 1990, 347, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.R.; Graceffa, P.; Dominguez, R. The crystal structure of uncomplexed actin in the adp state. Science 2001, 293, 708–711. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.Z.; Pollard, T.D. Mechanism of actin polymerization revealed by cryo-em structures of actin filaments with three different bound nucleotides. Proc. Natl. Acad. Sci. USA 2019, 116, 4265–4274. [Google Scholar] [CrossRef] [Green Version]
- Kotila, T.; Kogan, K.; Enkavi, G.; Guo, S.; Vattulainen, I.; Goode, B.L.; Lappalainen, P. Structural basis of actin monomer re-charging by cyclase-associated protein. Nat. Commun. 2018, 9, 1892. [Google Scholar] [CrossRef]
- Bubb, M.R.; Govindasamy, L.; Yarmola, E.G.; Vorobiev, S.M.; Almo, S.C.; Somasundaram, T.; Chapman, M.S.; Agbandje-McKenna, M.; McKenna, R. Polylysine induces an antiparallel actin dimer that nucleates filament assembly: Crystal structure at 3.5-a resolution. J. Biol. Chem. 2002, 277, 20999–21006. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ni, F.; Tian, X.; Kondrashkina, E.; Wang, Q.; Ma, J. Structural basis of actin filament nucleation by tandem w domains. Cell Rep. 2013, 3, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Mouilleron, S.; Wiezlak, M.; O’Reilly, N.; Treisman, R.; McDonald, N.Q. Structures of the phactr1 rpel domain and rpel motif complexes with g-actin reveal the molecular basis for actin binding cooperativity. Structure 2012, 20, 1960–1970. [Google Scholar] [CrossRef] [Green Version]
- Poukkula, M.; Kremneva, E.; Serlachius, M.; Lappalainen, P. Actin-depolymerizing factor homology domain: A conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton (Hoboken) 2011, 68, 471–490. [Google Scholar] [CrossRef] [PubMed]
- von der Ecken, J.; Muller, M.; Lehman, W.; Manstein, D.J.; Penczek, P.A.; Raunser, S. Structure of the f-actin-tropomyosin complex. Nature 2015, 519, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, F.; Pospich, S.; Funk, J.; Wagner, T.; Kullmer, F.; Arndt, H.D.; Bieling, P.; Raunser, S. Structural transitions of f-actin upon atp hydrolysis at near-atomic resolution revealed by cryo-em. Nat. Struct. Mol. Biol. 2018, 25, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Sasisekharan, V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968, 23, 283–438. [Google Scholar] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Pivovarova, A.V.; Khaitlina, S.Y.; Levitsky, D.I. Specific cleavage of the dnase-i binding loop dramatically decreases the thermal stability of actin. FEBS J. 2010, 277, 3812–3822. [Google Scholar] [CrossRef] [Green Version]
- Grintsevich, E.E.; Ge, P.; Sawaya, M.R.; Yesilyurt, H.G.; Terman, J.R.; Zhou, Z.H.; Reisler, E. Catastrophic disassembly of actin filaments via mical-mediated oxidation. Nat. Commun. 2017, 8, 2183. [Google Scholar] [CrossRef]
- Oda, T.; Iwasa, M.; Aihara, T.; Maeda, Y.; Narita, A. The nature of the globular- to fibrous-actin transition. Nature 2009, 457, 441–445. [Google Scholar] [CrossRef]
- Durer, Z.A.; Kudryashov, D.S.; Sawaya, M.R.; Altenbach, C.; Hubbell, W.; Reisler, E. Structural states and dynamics of the d-loop in actin. Biophys. J. 2012, 103, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Sano, K.; Maeda, K.; Oki, M.; Maeda, Y. Enhancement of protein expression in insect cells by a lobster tropomyosin cdna leader sequence. FEBS Lett. 2002, 532, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Spudich, J.A.; Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971, 246, 4866–4871. [Google Scholar] [PubMed]
- Mahaffy, R.E.; Pollard, T.D. Kinetics of the formation and dissociation of actin filament branches mediated by arp2/3 complex. Biophys. J. 2006, 91, 3519–3528. [Google Scholar] [PubMed] [Green Version]
- Kuhn, J.R.; Pollard, T.D. Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophys. J. 2005, 88, 1387–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, I.; Remmert, K.; Hammer, J.A., 3rd. Direct observation of the uncapping of capping protein-capped actin filaments by carmil homology domain 3. J. Biol. Chem. 2010, 285, 2707–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtemanche, N.; Pollard, T.D. Determinants of formin homology 1 (fh1) domain function in actin filament elongation by formins. J. Biol. Chem. 2012, 287, 7812–7820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, I.; Zweifel, M.E.; Courtemanche, N.; Pollard, T.D. Latrunculin a accelerates actin filament depolymerization in addition to sequestering actin monomers. Curr. Biol. 2018, 28, 3183–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasunaga, T.; Wakabayashi, T. Extensible and object-oriented system eos supplies a new environment for image analysis of electron micrographs of macromolecules. J. Struct. Biol. 1996, 116, 155–160. [Google Scholar] [CrossRef]
- Mizuno, H.; Tanaka, K.; Yamashiro, S.; Narita, A.; Watanabe, N. Helical rotation of the diaphanous-related formin mdia1 generates actin filaments resistant to cofilin. Proc. Natl. Acad. Sci. USA 2018, 115, E5000–E5007. [Google Scholar] [CrossRef] [Green Version]
- Oosawa, F.; Kasai, M. A theory of linear and helical aggregations of macromolecules. J. Mol. Biol. 1962, 4, 10–21. [Google Scholar] [CrossRef]
- Cooper, J.A.; Pollard, T.D. Methods to measure actin polymerization. Meth. Enzymol. 1982, 85 Pt B, 182–210. [Google Scholar]
- Kouyama, T.; Mihashi, K. Fluorimetry study of n-(1-pyrenyl)iodoacetamide-labelled f-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 1981, 114, 33–38. [Google Scholar] [CrossRef]
- Holmes, K.C.; Popp, D.; Gebhard, W.; Kabsch, W. Atomic model of the actin filament. Nature 1990, 347, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Rate constants for the reactions of atp- and adp-actin with the ends of actin filaments. J. Cell Biol. 1986, 103, 2747–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, I.; Vavylonis, D.; Pollard, T.D. Polymerization kinetics of adp- and adp-pi-actin determined by fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 8827–8832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of f-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Narita, A. Adf/cofilin regulation from a structural viewpoint. J. Muscle Res. Cell Motil. 2020, 41, 141–151. [Google Scholar] [CrossRef]
- Kanellos, G.; Frame, M.C. Cellular functions of the adf/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, I.; Takeda, S.; Oda, T.; Honda, H.; Narita, A.; Maeda, Y. Polymerization and depolymerization of actin with nucleotide states at filament ends. Biophys. Rev. 2018, 10, 1513–1519. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Carlier, M.F.; Pantaloni, D. Control of actin assembly dynamics in cell motility. J. Biol. Chem. 2007, 282, 23005–23009. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | Mutant | |||
|---|---|---|---|---|
| B-end | P-end | B-end | P-end | |
| k+ (µM−1 s−1) a | 11.0 ± 0.8 c | 1.1 ± 0.2 | 7.1 ± 0.6 | 0.7 ± 0.1 |
| k− (s−1) | 1.8 ± 0.3 | 0.19 ± 0.08 | 0.6 ± 0.2 | 0.13 ± 0.06 |
| Cc (µM) b | 0.17 ± 0.03 | 0.18 ± 0.09 | 0.09 ± 0.03 | 0.2 ± 0.1 |
| N d | 46 | 56 | 52 | 50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuzaki, M.; Fujiwara, I.; Kashima, S.; Matsumoto, T.; Oda, T.; Hayashi, M.; Maeda, K.; Takiguchi, K.; Maéda, Y.; Narita, A. D-Loop Mutation G42A/G46A Decreases Actin Dynamics. Biomolecules 2020, 10, 736. https://doi.org/10.3390/biom10050736
Matsuzaki M, Fujiwara I, Kashima S, Matsumoto T, Oda T, Hayashi M, Maeda K, Takiguchi K, Maéda Y, Narita A. D-Loop Mutation G42A/G46A Decreases Actin Dynamics. Biomolecules. 2020; 10(5):736. https://doi.org/10.3390/biom10050736
Chicago/Turabian StyleMatsuzaki, Mizuki, Ikuko Fujiwara, Sae Kashima, Tomoharu Matsumoto, Toshiro Oda, Masahito Hayashi, Kayo Maeda, Kingo Takiguchi, Yuichiro Maéda, and Akihiro Narita. 2020. "D-Loop Mutation G42A/G46A Decreases Actin Dynamics" Biomolecules 10, no. 5: 736. https://doi.org/10.3390/biom10050736