The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives




Abstract
:1. Introduction
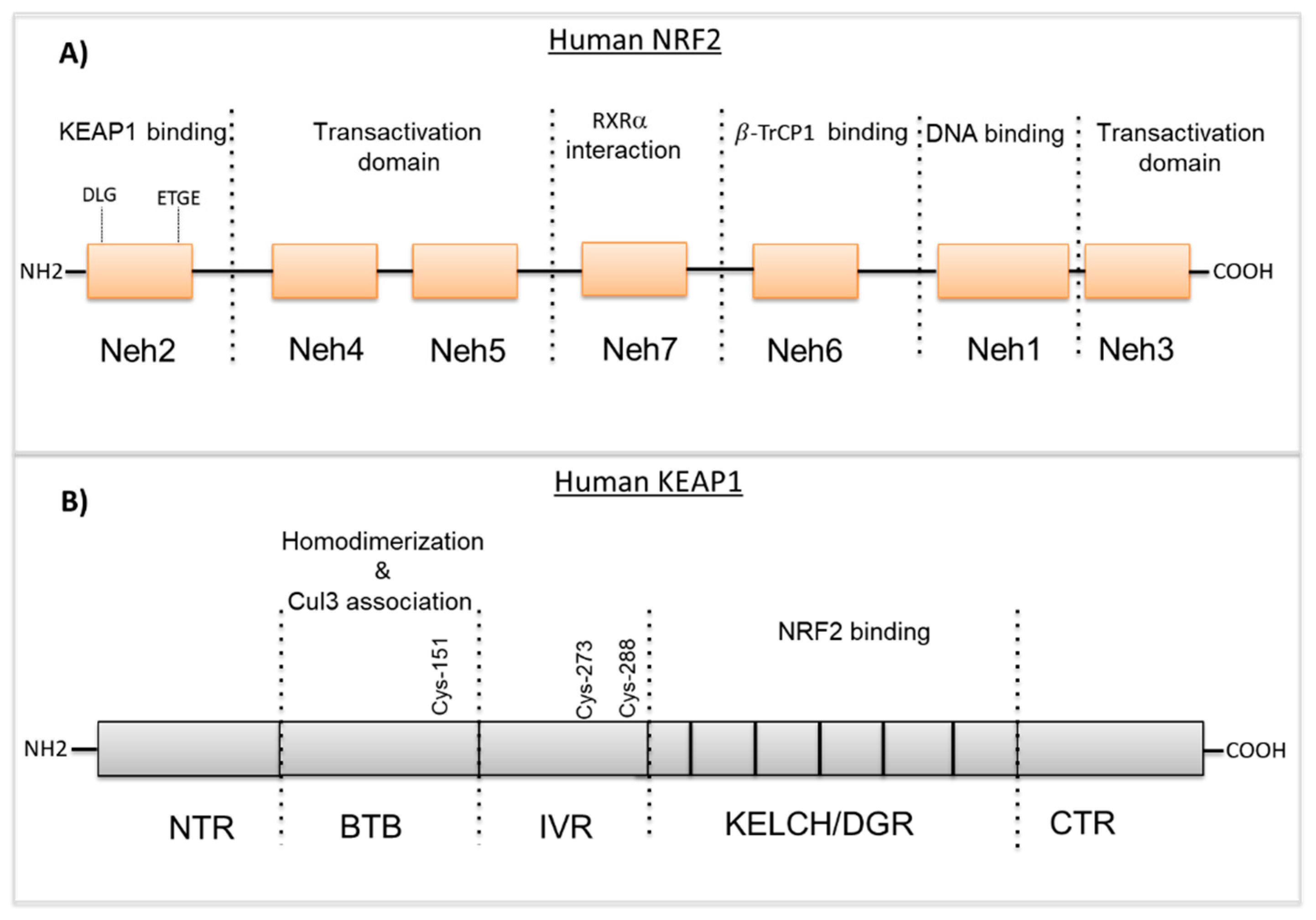
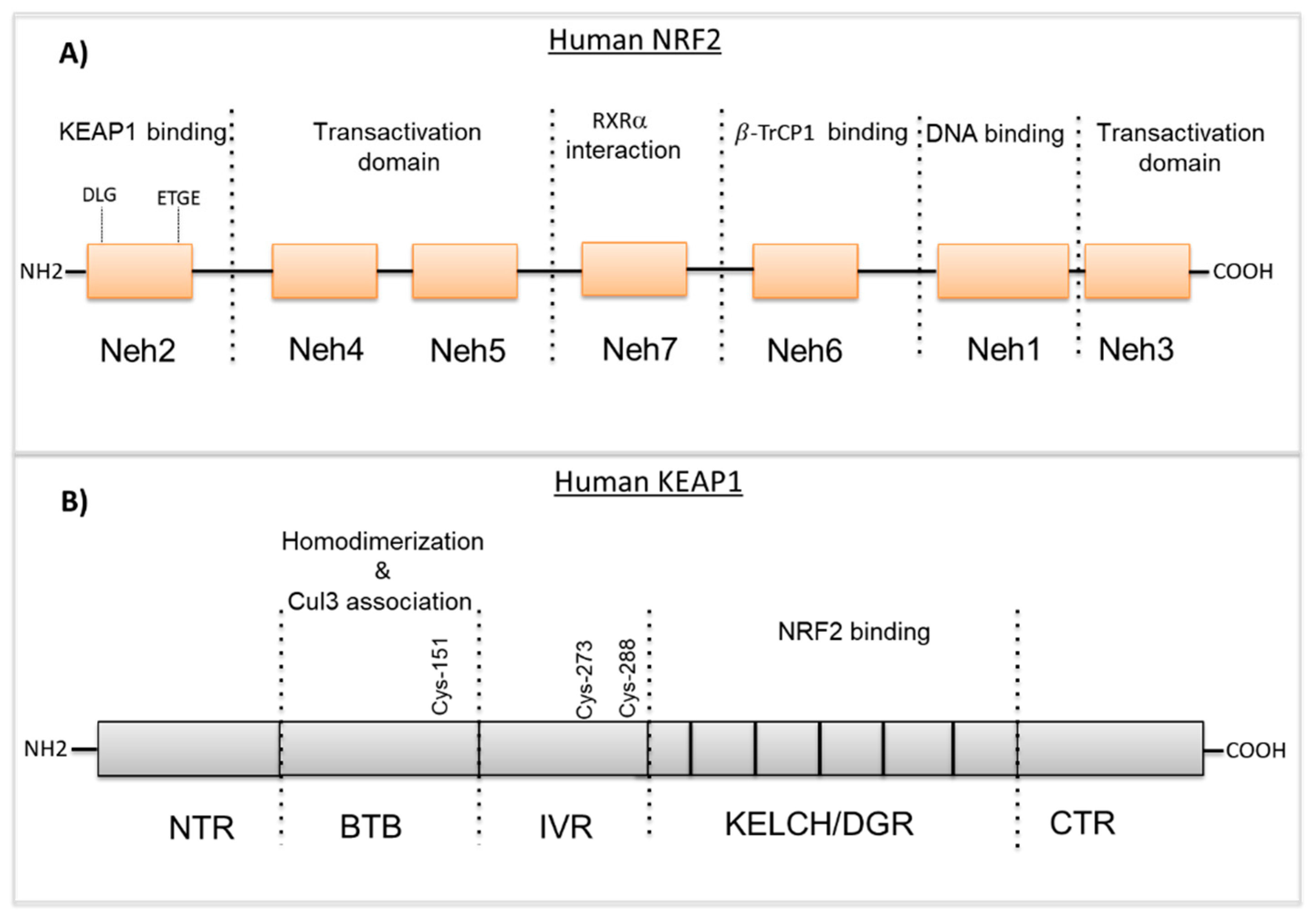
2. NRF2 and KEAP1 Structure and Function
2.1. NRF2 Structure
2.2. KEAP1 Structure
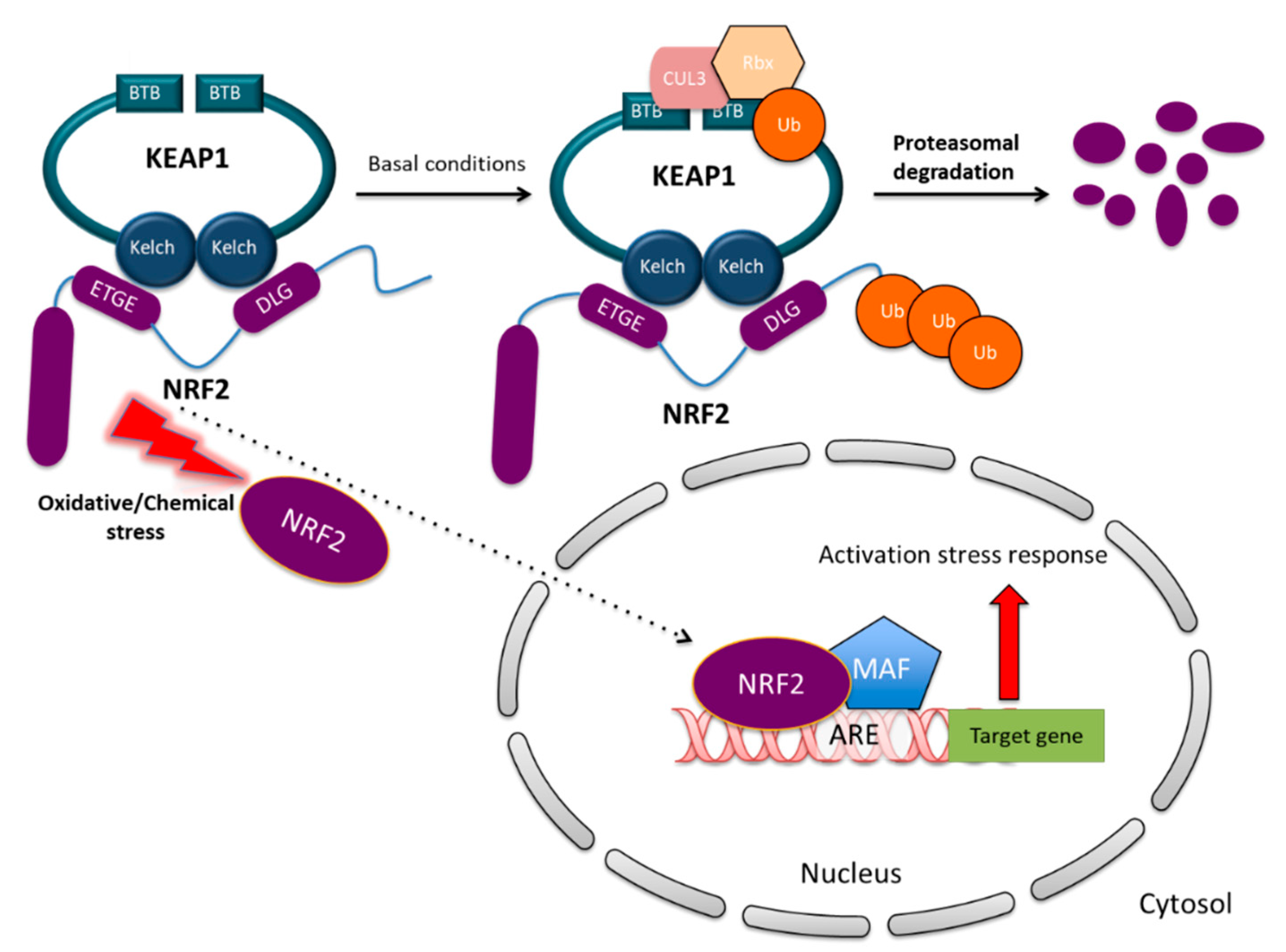
2.3. NRF2 and KEAP1 Regulation under Normal and Stressed Conditions
2.4. The Transcriptional Program Elicited by NRF2 Activation and Its Biological Effects
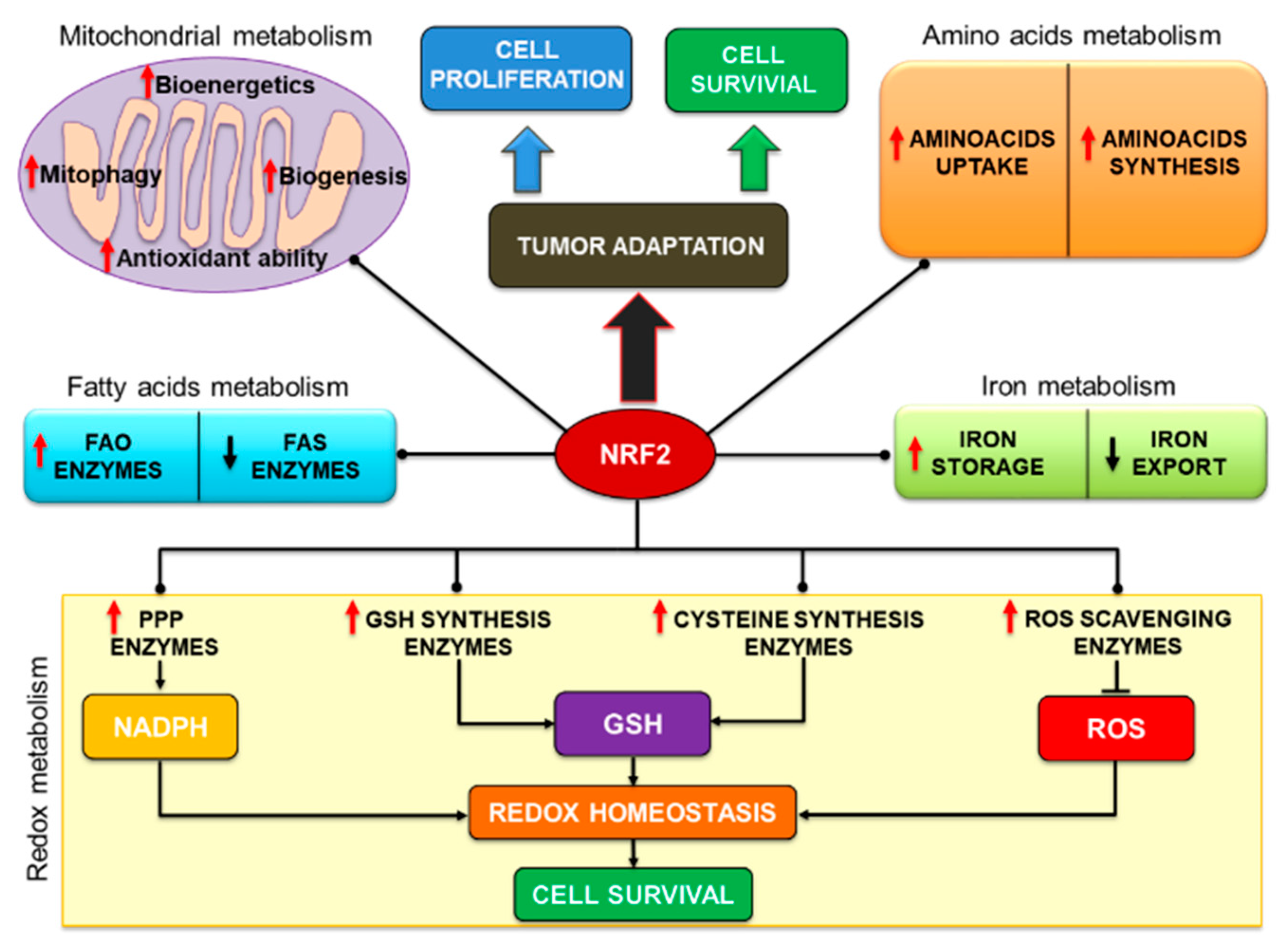
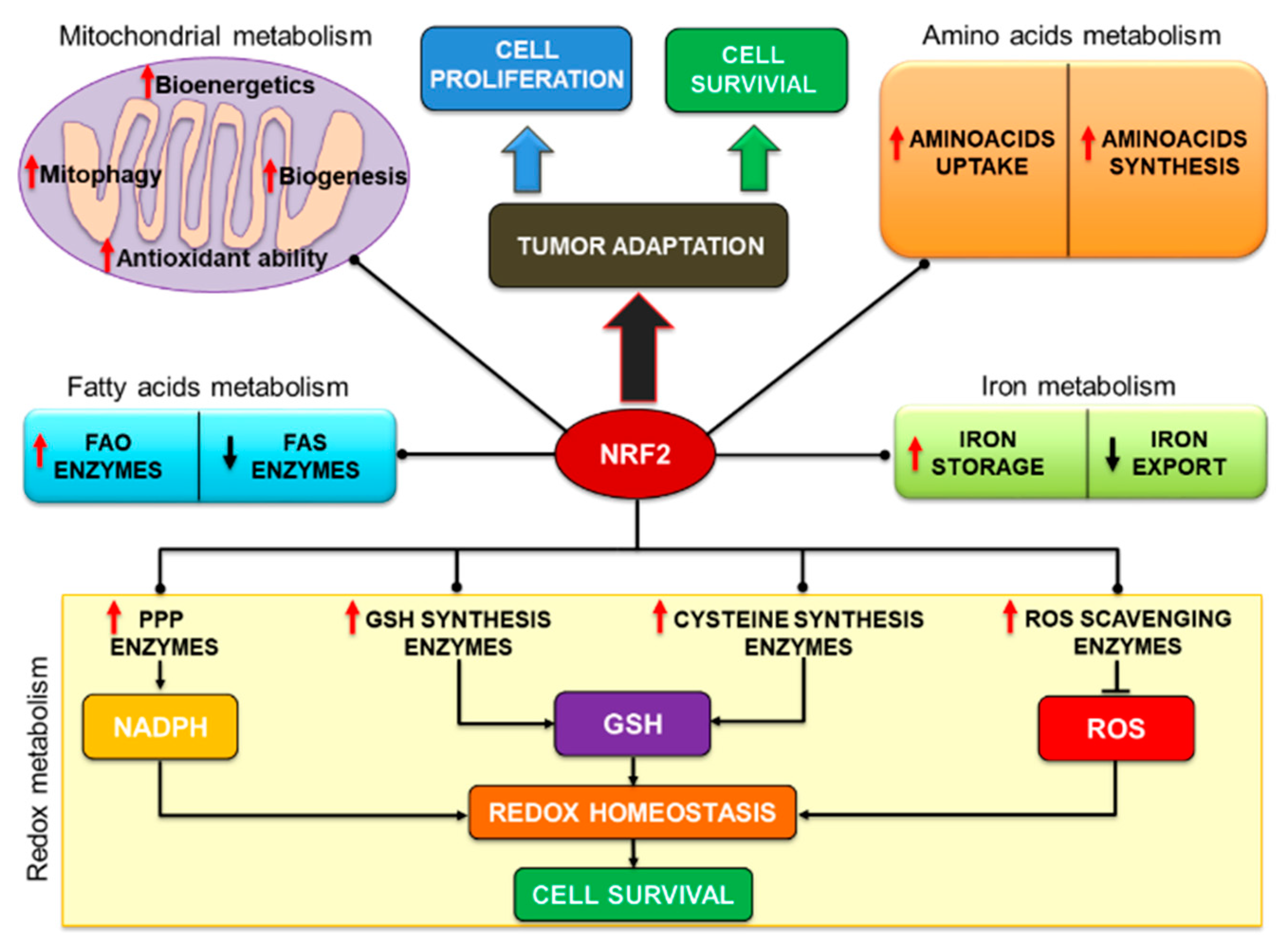
3. Role of the NRF2/KEAP1 Pathway in Tumor Metabolism
3.1. NRF2 Controls Mitochondrial Function Linking Metabolism to Redox Balance
3.1.1. NRF2 Regulates Mitochondrial Biogenesis, Turnover and Mitochondrial Network Dynamics
3.1.2. NRF2 Regulates Mitochondrial Respiration and Redox Homeostasis
3.2. NRF2 Regulates Fatty Acids Metabolism
3.3. NRF2 Regulates Aminoacids Metabolism
3.3.1. NRF2 Controls Aminoacids Uptake and Biosynthesis to Support Proliferation and Survival
3.3.2. NRF2 Controls xCT Antiport to Support Cell Survival Leading to Metabolic Addiction
3.3.3. NRF2 Regulates Cysteine Biosynthesis and Metabolic Transformation
3.4. NRF2 Is Regulated by H2S Metabolism
3.5. NRF2 Controls Iron Metabolism
3.6. NRF2 Controls Redox Homeostasis through NADPH Synthesis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vomund, S.; Schafer, A.; Parnham, M.J.; Brune, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namani, A.; Li, Y.; Wang, X.J.; Tang, X. Modulation of NRF2 signaling pathway by nuclear receptors: Implications for cancer. Biochim. Biophys. Acta 2014, 1843, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. Pharmacological Applications of Nrf2 Inhibitors as Potential Antineoplastic Drugs. Int J. Mol. Sci. 2019, 20, 2025. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintard, L.; Willems, A.; Peter, M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. Embo J. 2004, 23, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Lo, S.C.; Sun, Z.; Habib, G.M.; Lieberman, M.W.; Hannink, M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J. Biol. Chem. 2005, 280, 30091–30099. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Surh, Y.J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005, 224, 171–184. [Google Scholar] [CrossRef]
- Cleasby, A.; Yon, J.; Day, P.J.; Richardson, C.; Tickle, I.J.; Williams, P.A.; Callahan, J.F.; Carr, R.; Concha, N.; Kerns, J.K.; et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS ONE 2014, 9, e98896. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Rojo de la Vega, M.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 293, 2029–2040. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Telakowski-Hopkins, C.A.; King, R.G.; Pickett, C.B. Glutathione S-transferase Ya subunit gene: Identification of regulatory elements required for basal level and inducible expression. Proc. Natl. Acad. Sci. USA 1988, 85, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Buha, A.; Telkoparan-Akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential Applications of NRF2 Modulators in Cancer Therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Chanas, S.A.; Jiang, Q.; McMahon, M.; McWalter, G.K.; McLellan, L.I.; Elcombe, C.R.; Henderson, C.J.; Wolf, C.R.; Moffat, G.J.; Itoh, K.; et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002, 365, 405–416. [Google Scholar] [CrossRef]
- Moinova, H.R.; Mulcahy, R.T. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem. Biophys. Res. Commun. 1999, 261, 661–668. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, A.; Nishimoto, M.; Himeno, S.; Imura, N.; Tsujimoto, M.; Kunimoto, M.; Hara, S. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: Role of NF-E2-related factor-2. J. Cell Physiol. 2005, 203, 529–537. [Google Scholar] [CrossRef]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef]
- Hayes, J.D.; Ashford, M.L. Nrf2 orchestrates fuel partitioning for cell proliferation. Cell Metab. 2012, 16, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, J.M.; Dieter, M.Z.; Aleksunes, L.M.; Slitt, A.L.; Guo, G.; Tanaka, Y.; Scheffer, G.L.; Chan, J.Y.; Manautou, J.E.; Chen, Y.; et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology 2007, 46, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Wible, R.S.; Ramanathan, C.; Sutter, C.H.; Olesen, K.M.; Kensler, T.W.; Liu, A.C.; Sutter, T.R. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. Elife 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.J.; Dudakov, J.A.; Takahashi, K.; Shieh, J.H.; Velardi, E.; Holland, A.M.; Singer, N.V.; West, M.L.; Smith, O.M.; Young, L.F.; et al. Nrf2 regulates haematopoietic stem cell function. Nat. Cell Biol. 2013, 15, 309–316. [Google Scholar] [CrossRef]
- Jang, J.; Wang, Y.; Lalli, M.A.; Guzman, E.; Godshalk, S.E.; Zhou, H.; Kosik, K.S. Primary Cilium-Autophagy-Nrf2 (PAN) Axis Activation Commits Human Embryonic Stem Cells to a Neuroectoderm Fate. Cell 2016, 165, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, P.C.; Slentz, D.H.; Pisitkun, T.; Saeed, F.; Hoffert, J.D.; Knepper, M.A. Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J. Am. Soc. Nephrol. 2013, 24, 1793–1805. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, H.; Cordova, M.; Chen, C.J.; Rajadhyaksha, M. Confocal imaging-guided laser ablation of basal cell carcinomas: An ex vivo study. J. Investig. Derm. 2015, 135, 612–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Hu, B.; Zang, F.; Wang, J.; Zhang, X.; Chen, H. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell Death Dis. 2019, 10, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, A.K.; McMahon, M.; Plummer, S.M.; Higgins, L.G.; Penning, T.M.; Igarashi, K.; Hayes, J.D. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: Demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009, 30, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Jung, K.A.; Choi, B.H.; Nam, C.W.; Song, M.; Kim, S.T.; Lee, J.Y.; Kwak, M.K. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol. Lett. 2013, 218, 39–49. [Google Scholar] [CrossRef]
- Kuda, O.; Brezinova, M.; Silhavy, J.; Landa, V.; Zidek, V.; Dodia, C.; Kreuchwig, F.; Vrbacky, M.; Balas, L.; Durand, T.; et al. Nrf2-Mediated Antioxidant Defense and Peroxiredoxin 6 Are Linked to Biosynthesis of Palmitic Acid Ester of 9-Hydroxystearic Acid. Diabetes 2018, 67, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Aguilar-Bryan, L.; Vaxillaire, M.; Dechaume, A.; Wang, Y.; Dean, M.; Moitra, K.; Bryan, J.; Schuetz, J.D. Conserved intramolecular disulfide bond is critical to trafficking and fate of ATP-binding cassette (ABC) transporters ABCB6 and sulfonylurea receptor 1 (SUR1)/ABCC8. J. Biol. Chem. 2011, 286, 8481–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.K.; Kwon, S.W.; Han, D.H.; Lee, Y.H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Inose-Maruyama, A.; Taniuchi, S.; Kosaka, K.; Yoshida, H.; Yamazaki, H.; Kasai, S.; Harada, N.; Kaufman, R.J.; Oyadomari, S.; et al. Concomitant Nrf2- and ATF4-activation by Carnosic Acid Cooperatively Induces Expression of Cytoprotective Genes. Int J. Mol. Sci. 2019, 20, 1706. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Iida, K.; Kang, M.I.; Kobayashi, A.; Mizukami, M.; Tong, K.I.; McMahon, M.; Hayes, J.D.; Itoh, K.; Yamamoto, M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 2005, 433, 342–350. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Riis, S.; Murray, J.B.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3beta-Nrf2-BNIP3 Pathway. Cells 2020, 9, 147. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Joe, Y.; Zheng, M.; Kim, H.J.; Yu, J.K.; Cho, G.J.; Chang, K.C.; Kim, H.K.; Han, J.; Ryter, S.W.; et al. Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production. Antioxid. Redox Signal. 2014, 20, 2589–2605. [Google Scholar] [CrossRef] [Green Version]
- Shukla, K.; Sonowal, H.; Saxena, A.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibitor, fidarestat regulates mitochondrial biogenesis via Nrf2/HO-1/AMPK pathway in colon cancer cells. Cancer Lett. 2017, 411, 57–63. [Google Scholar] [CrossRef]
- Negrette-Guzman, M.; Huerta-Yepez, S.; Vega, M.I.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Medina-Campos, O.N.; Rodriguez, E.; Tapia, E.; Pedraza-Chaverri, J. Sulforaphane induces differential modulation of mitochondrial biogenesis and dynamics in normal cells and tumor cells. Food Chem. Toxicol. 2017, 100, 90–102. [Google Scholar] [CrossRef]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under Oxidative Stress Conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef]
- Castrejon-Jimenez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernandez-Perez, A.D.; Luna-Angulo, A.B.; Chacon-Salinas, R.; Coral-Vazquez, R.M.; Estrada-Garcia, I.; et al. Ursolic and Oleanolic Acids Induce Mitophagy in A549 Human Lung Cancer Cells. Molecules 2019, 24, 3444. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Sun, W.; Wang, X.; Long, J.; Liu, X.; Feng, Z.; Liu, J. Punicalagin attenuates palmitate-induced lipotoxicity in HepG2 cells by activating the Keap1-Nrf2 antioxidant defense system. Mol. Nutr. Food Res. 2016, 60, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Liu, Z.; Wang, L.; Shi, R.; Chu, C.; Xiang, M.; Tian, Q.; Liu, X. Protective effects of lipoic acid against acrylamide-induced neurotoxicity: Involvement of mitochondrial energy metabolism and autophagy. Food Funct. 2017, 8, 4657–4667. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.A.; Lee, S.; Kwak, M.K. NFE2L2/NRF2 Activity Is Linked to Mitochondria and AMP-Activated Protein Kinase Signaling in Cancers Through miR-181c/Mitochondria-Encoded Cytochrome c Oxidase Regulation. Antioxid. Redox Signal. 2017, 27, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of Glutathione Metabolism in IDH1-Mutated Glioma. Mol. Cancer 2020, 19, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Dong, J.L.; Chen, Y.L.; Liu, Y.; Huang, S.S.; Zhong, X.L.; Cheng, Y.H.; Wang, Z.G. Nrf2 mediates the protective effects of homocysteine by increasing the levels of GSH content in HepG2 cells. Mol. Med. Rep. 2017, 16, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Sulik, K.K.; Chen, S.Y. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, T.; Shafer, J.; Fiskum, G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic. Biol. Med. 2011, 51, 2164–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Wu, Y.; Liu, J.; Shen, X.; Tong, F.; Xu, G.; Shen, R. GSK-3beta Inhibitor Induces Expression of Nrf2/TrxR2 Signaling Pathway to Protect against Renal Ischemia/Reperfusion Injury in Diabetic Rats. Kidney Blood Press Res. 2016, 41, 937–946. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Kondo, H.; Tawara, A.; Sasaguri, Y.; Kohno, K. Quercetin induces the expression of peroxiredoxins 3 and 5 via the Nrf2/NRF1 transcription pathway. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Festa, F.; Freet, C.S.; Lee, J.P.; Hirschler-Laszkiewicz, I.M.; Chen, S.J.; Keefer, K.A.; Wang, H.G.; Patterson, A.D.; Cheung, J.Y.; et al. The Human Transient Receptor Potential Melastatin 2 Ion Channel Modulates ROS Through Nrf2. Sci. Rep. 2019, 9, 14132. [Google Scholar] [CrossRef] [Green Version]
- Negrette-Guzman, M.; Huerta-Yepez, S.; Tapia, E.; Pedraza-Chaverri, J. Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: A seemingly contradictory dual role and an integrative hypothesis. Free Radic Biol. Med. 2013, 65, 1078–1089. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.; Lynn, D.A.; Lo, J.Y.; Paek, J.; Curran, S.P. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat. Commun. 2014, 5, 5048. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Yang, X.; Yuan, F.; Zhang, L.; Wang, Y.; Wang, L.; Mao, Z.; Luo, J.; Zhang, H.; Zhu, W.G.; et al. Increased Amino Acid Uptake Supports Autophagy-Deficient Cell Survival upon Glutamine Deprivation. Cell Rep. 2018, 23, 3006–3020. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.F.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouyssegur, J.; Vucetic, M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [Green Version]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef] [Green Version]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [Green Version]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. Embo. J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020, 31, 339–350.e4. [Google Scholar] [CrossRef] [PubMed]
- Averous, J.; Bruhat, A.; Jousse, C.; Carraro, V.; Thiel, G.; Fafournoux, P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J. Biol. Chem. 2004, 279, 5288–5297. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Xiong, Z.; Huang, C.; Li, J.; Yang, W.; Han, Y.; Paiboonrungruan, C.; Major, M.B.; Chen, K.N.; Kang, X.; et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019, 294, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 2019, 8. [Google Scholar] [CrossRef]
- Zhu, J.; Berisa, M.; Schworer, S.; Qin, W.; Cross, J.R.; Thompson, C.B. Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab. 2019, 30, 865–876.e5. [Google Scholar] [CrossRef]
- Akbari, M.; Sogutdelen, E.; Juriasingani, S.; Sener, A. Hydrogen Sulfide: Emerging Role in Bladder, Kidney, and Prostate Malignancies. Oxid. Med. Cell Longev. 2019, 2019, 2360945. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Szabo, C. Hydrogen Sulfide and Cancer. Handb. Exp. Pharm. 2015, 230, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.; Stern, A.; Monteiro, H.P. S-nitrosothiols and H2S donors: Potential chemo-therapeutic agents in cancer. Redox Biol. 2019, 27, 101190. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide exerts a protective effect against cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. Febs Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, L.; Junhua, W.; Long, L.; Jun, Y.; Yang, X. Exogenous Hydrogen Sulfide Protects SH-SY5Y Cells from OGD/RInduced Injury. Curr. Mol. Med. 2017, 17, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Wei, Z.; Tao, L.; Wang, S.; Zhang, F.; Shen, C.; Wu, H.; Liu, Z.; Zhu, P.; Wang, A.; et al. Prophylaxis of Diallyl Disulfide on Skin Carcinogenic Model via p21-dependent Nrf2 stabilization. Sci. Rep. 2016, 6, 35676. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.Y.; Zhu, X.S.; Xu, H.Y.; Zhao, Z.X.; Li, S.Y.; Li, S.Z.; Cai, J.H.; Cao, J.M. Diallyl trisulfide suppresses tumor growth through the attenuation of Nrf2/Akt and activation of p38/JNK and potentiates cisplatin efficacy in gastric cancer treatment. Acta Pharm. Sin. 2017, 38, 1048–1058. [Google Scholar] [CrossRef]
- Wang, R.; Tao, B.; Fan, Q.; Wang, S.; Chen, L.; Zhang, J.; Hao, Y.; Dong, S.; Wang, Z.; Wang, W.; et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine 2019, 45, 108–123. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Mertens, C.; Tomat, E.; Brune, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 2005, 24, 7567–7578. [Google Scholar] [CrossRef] [Green Version]
- Kweon, M.H.; Adhami, V.M.; Lee, J.S.; Mukhtar, H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J. Biol. Chem. 2006, 281, 33761–33772. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, S.; Wang, X.; Guo, W.; Wang, L.; Zhang, D.; Yuan, L.; Zhang, Z.; Xu, Y.; Liu, S. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell. Signal. 2015, 27, 168–176. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, C.; Shi, Y.; Lu, H.; Xu, R.; He, X. Nuclear transcription factor Nrf2 suppresses prostate cancer cells growth and migration through upregulating ferroportin. Oncotarget 2016, 7, 78804–78812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Vashisht, A.A.; Liang, B.X.; Duckworth, S.J.; Praslicka, B.J.; Wohlschlegel, J.A.; Ooi, A. Fumarate Mediates a Chronic Proliferative Signal in Fumarate Hydratase-Inactivated Cancer Cells by Increasing Transcription and Translation of Ferritin Genes. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Bao, L.; Zhang, Z.; Yi, X. Nrf2 induces cisplatin resistance via suppressing the iron export related gene SLC40A1 in ovarian cancer cells. Oncotarget 2017, 8, 93502–93515. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Campbell, M.R.; Karaca, M.; Adamski, K.N.; Chorley, B.N.; Wang, X.; Bell, D.A. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid. Med. Cell Longev. 2013, 2013, 120305. [Google Scholar] [CrossRef] [Green Version]
- Carter, D.R.; Sutton, S.K.; Pajic, M.; Murray, J.; Sekyere, E.O.; Fletcher, J.; Beckers, A.; De Preter, K.; Speleman, F.; George, R.E.; et al. Glutathione biosynthesis is upregulated at the initiation of MYCN-driven neuroblastoma tumorigenesis. Mol. Oncol. 2016, 10, 866–878. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hlouschek, J.; Ritter, V.; Wirsdorfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, B.; Moller, T.; Hu-Moore, K.; Carrington, S.; Faull, K.F.; Sen, S.; Mascharak, P.K. Attenuation of Antioxidant Capacity in Human Breast Cancer Cells by Carbon Monoxide through Inhibition of Cystathionine beta-Synthase Activity: Implications in Chemotherapeutic Drug Sensitivity. J. Med. Chem. 2017, 60, 8000–8010. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, G.R.; Kim, J.; Baek, J.Y.; Jo, Y.J.; Hong, S.E.; Kim, S.H.; Lee, J.; Lee, H.I.; Park, S.K.; et al. Sulfiredoxin inhibitor induces preferential death of cancer cells through reactive oxygen species-mediated mitochondrial damage. Free Radic. Biol. Med. 2016, 91, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Xu, I.M.; Lai, R.K.; Lin, S.H.; Tse, A.P.; Chiu, D.K.; Koh, H.Y.; Law, C.T.; Wong, C.M.; Cai, Z.; Wong, C.C.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, e2213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.S.; Zhang, Z.G.; Du, G.Y.; Sun, H.L.; Liu, H.Y.; Zhou, Z.; Gou, X.M.; Wu, X.H.; Yu, X.Y.; Huang, Y.H. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Badur, M.G.; Luebeck, J.; Magana, J.H.; Birmingham, A.; Sasik, R.; Ahn, C.S.; Ideker, T.; Metallo, C.M.; Mali, P. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol. Cell 2018, 69, 699–708.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, M.; Yang, L.; Liao, H.; Liang, X.; Xie, B.; Xiong, J.; Tao, X.; Chen, X.; Cheng, Y.; Chen, X.; et al. Metformin sensitizes endometrial cancer cells to chemotherapy through IDH1-induced Nrf2 expression via an epigenetic mechanism. Oncogene 2018, 37, 5666–5681. [Google Scholar] [CrossRef]
- Lee, D.; Xu, I.M.; Chiu, D.K.; Lai, R.K.; Tse, A.P.; Lan Li, L.; Law, C.T.; Tsang, F.H.; Wei, L.L.; Chan, C.Y.; et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J. Clin. Investig. 2017, 127, 1856–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Cheng, X.D.; Zhang, J.; Zhang, W.D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 121. [Google Scholar] [CrossRef] [Green Version]
- Nam, L.B.; Keum, Y.S. Binding partners of NRF2: Functions and regulatory mechanisms. Arch. Biochem. Biophys. 2019, 678, 108184. [Google Scholar] [CrossRef]
- Takahashi, T.; Sonobe, M.; Menju, T.; Nakayama, E.; Mino, N.; Iwakiri, S.; Nagai, S.; Sato, K.; Miyahara, R.; Okubo, K.; et al. Mutations in Keap1 are a potential prognostic factor in resected non-small cell lung cancer. J. Surg. Oncol. 2010, 101, 500–506. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008, 135, 1358–1368.e4. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, I.S.; Maksim, N.J.; Amouzougan, E.A.; Gallion, B.W.; Raviele, A.L.; Ooi, A. Sustained NRF2 activation in hereditary leiomyomatosis and renal cell cancer (HLRCC) and in hereditary tyrosinemia type 1 (HT1). Biochem. Soc. Trans. 2015, 43, 650–656. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Li, L.; Shen, C.; Nakamura, E.; Ando, K.; Signoretti, S.; Beroukhim, R.; Cowley, G.S.; Lizotte, P.; Liberzon, E.; Bair, S.; et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 2013, 24, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.L.; Murray, J.B.; Park, B.K.; Copple, I.M. Roles of Nrf2 in drug and chemical toxicity. Curr. Opin. Toxicol. 2016, 1, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Huang, M.T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.N. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Biochemical Function | Gene Symbol | Extended Name | References Number |
|---|---|---|---|
| Antioxidants | GCLC | Glutamate–cysteine ligase, catalytic subunit | [30,50] |
| GCLM | Glutamate–cysteine ligase, modifier subunit | [25,45] | |
| GPX1,2,4 | Glutathione peroxidase 1 | [25] | |
| GSR1 | Glutathione reductase 1 | [51] | |
| NQO1 | NAD(P)H:quinoneo xidoreductase 1 | [51] | |
| SLC7A11 | Sodium-independent cysteine-glutamate antiporter | [52] | |
| SRXN1 | Sulfiredoxin 1 | [52] | |
| PRDX1 | Peroxiredoxin 1 | [53] | |
| TXN1 | Thioredoxin | [28,30] | |
| TXNRD1 | Thioredoxin reductase 1 | [30] | |
| Phase I detoxification | ADH7 | Alcohol dehydrogenase class 4 mu/sigma chain | [40] |
| AKR1B1, | Aldo-keto reductase family 1 member B1 | [23] | |
| AKR1B8 | Aldo-keto reductase family 1. member B8 | [23] | |
| AKR1B10 | Aldo-keto reductase family 1. member B10 | [23] | |
| AKR1CL | Aldo-keto reductase family 1. member C-like | [23] | |
| ALDH1A1 | Aldehyde dehydrogenase 1 family member A1 | [54] | |
| ALDH3A1 | Aldehyde dehydrogenase 3 family member A1 | [23] | |
| CBR1 | Carbonyl reductase 1 | [46] | |
| CYP1B1 | Cytochrome P450 | [40] | |
| PTGR1 | Prostaglandin reductase 1 | [40] | |
| EPXH1 | Epoxide hydrolase 1, microsomal | [40] | |
| Phase II detoxification | GSTA1,2 | Glutathione S-transferase alpha 1,253,4 | [26] |
| GSTM1,2,3,4 | Glutathione S-transferase mu 1 | [26] | |
| MGST1 | Microsomal glutathione S-transferase | [55] | |
| UGT1A1 | UDP Glucuronosyltransferase 1 | [46] | |
| UGT1A2 | UDP glucuronosyltransferase 1 family. polypeptide A2 | [46] | |
| Phase III detoxification | ABCB6 | ATP-binding cassette, subfamily B (MDR/Tap) member 6 | [56] |
| ABCC1 | ATP-binding cassette, subfamily C(CFTR/MRP) | [34] | |
| ABCC2 | ATP-binding cassette, subfamily C(CFTR/MRP) | [34] | |
| ABCC3 | ATP-binding cassette, subfamily C(CFTR/MRP) | [34] | |
| ABCC4 | ATP-binding cassette, subfamily C(CFTR/MRP) | [34] | |
| ABCC5 | ATP-binding cassette, subfamily C(CFTR/MRP) | [33] | |
| Heme and iron metabolism | FTH1 | Ferritin heavy chain 1 | [48] |
| FTL1 | Ferritin light chain 1 | [46] | |
| HMOX1 | Heme oxygenase 1 | [32] | |
| NADPH generation | G6PD | Glucose-6-phosphate dehydrogenase | [31] |
| IDH1 | NADP-dependent isocitrate dehydrogenase | [31] | |
| PGD | 6-phosphogluconate dehydrogenase | [31] | |
| ME1 | Malic enzyme 1 | [31] | |
| Apoptosis | BCL2 | B-cell lymphoma 2 | [44] |
| Autophagy | ATG5 | Autophagy protein 5 | [48,57] |
| ATG7 | Autophagy protein 7 | [48,57] | |
| LC3B | Microtubule-associated protein 1A/1B-light chain 3B | [43,52] | |
| ULK1 | UNC-51 autophagy-activating kinase 1 | [58] | |
| Proteasomal degradation and unfolded protein response | ATF4 | Activating transcription factor-4 | [59,60] |
| PSMA1 | Proteasome subunit alpha type-1 | [50] | |
| PSMB5 | Proteasome subunit beta type-5 | [50] | |
| PSMC1 | Proteasome AAA-ATPase subunit Rpt2 | [50] | |
| SQSTM1 | Sequestosome 1 (p62) | [61] | |
| Regulation of NRF2 signaling | KEAP1 | Kelch-like ECH-associated protein 1 | [62] |
| NFE2L2 | Nuclear factor, erythroid 2-like 2 (NRF2) | [63,64] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules 2020, 10, 791. https://doi.org/10.3390/biom10050791
Panieri E, Telkoparan-Akillilar P, Suzen S, Saso L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules. 2020; 10(5):791. https://doi.org/10.3390/biom10050791
Chicago/Turabian StylePanieri, Emiliano, Pelin Telkoparan-Akillilar, Sibel Suzen, and Luciano Saso. 2020. "The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives" Biomolecules 10, no. 5: 791. https://doi.org/10.3390/biom10050791
APA StylePanieri, E., Telkoparan-Akillilar, P., Suzen, S., & Saso, L. (2020). The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules, 10(5), 791. https://doi.org/10.3390/biom10050791