Yeast Sphingolipid-Enriched Domains and Membrane Compartments in the Absence of Mannosyldiinositolphosphorylceramide

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Strains
2.2. Media and Growth Conditions
2.3. Plasma Membrane Isolation
2.4. Lipid Extraction
2.5. Giant Unilamellar Vesicles (GUVs) Preparation
2.6. Fluorescence Spectroscopy Measurements and Data Analysis
2.7. Fluorescence Intensity and Lifetime Imaging by Confocal Microscopy
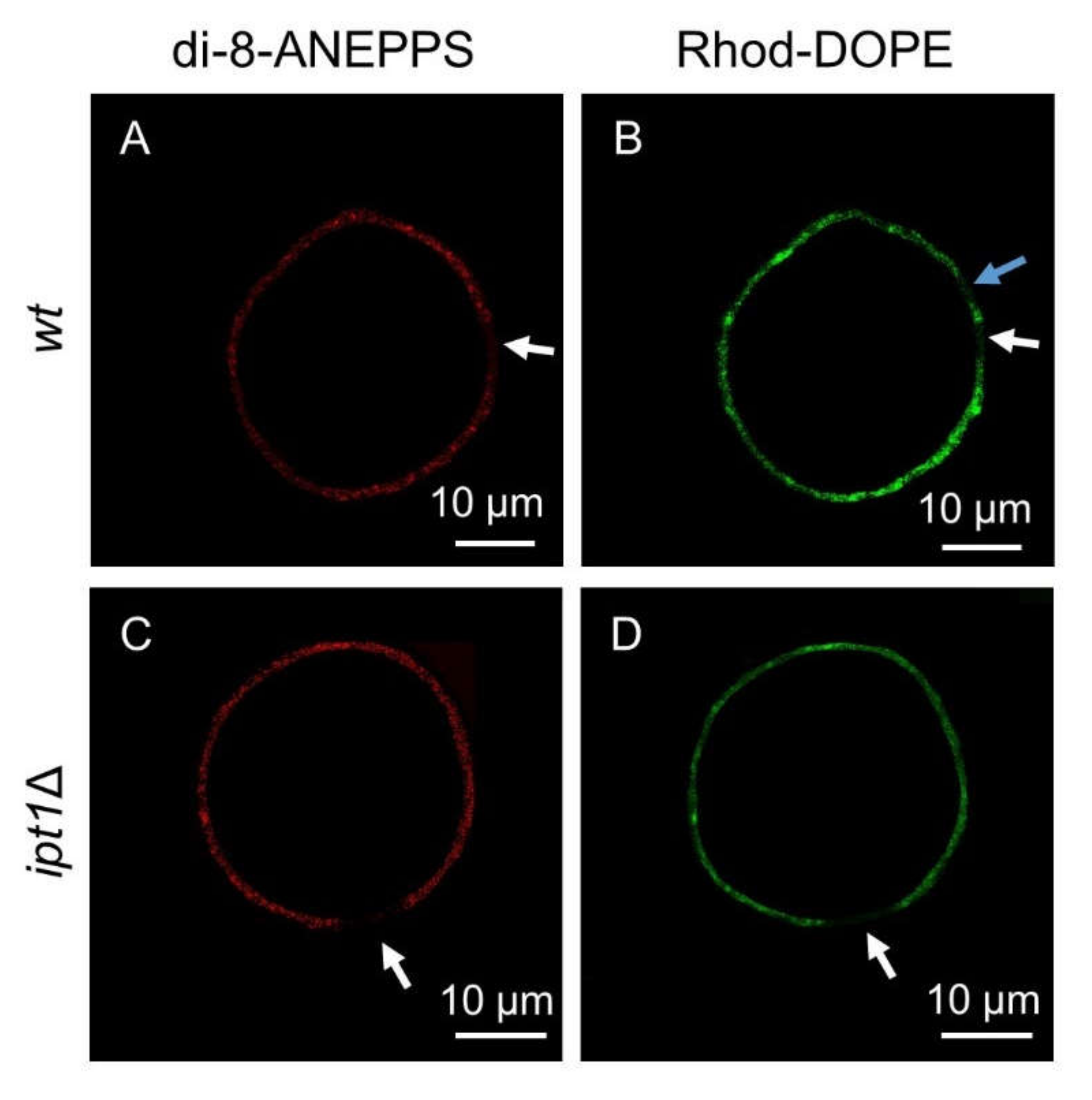
2.7.1. GUV and Living Cells Labeled with Di-8-ANEPPS or Rhod-DOPE
2.7.2. Yeast Living Cells Tagged with GFP and mRFP
2.8. Statistical Analysis
3. Results
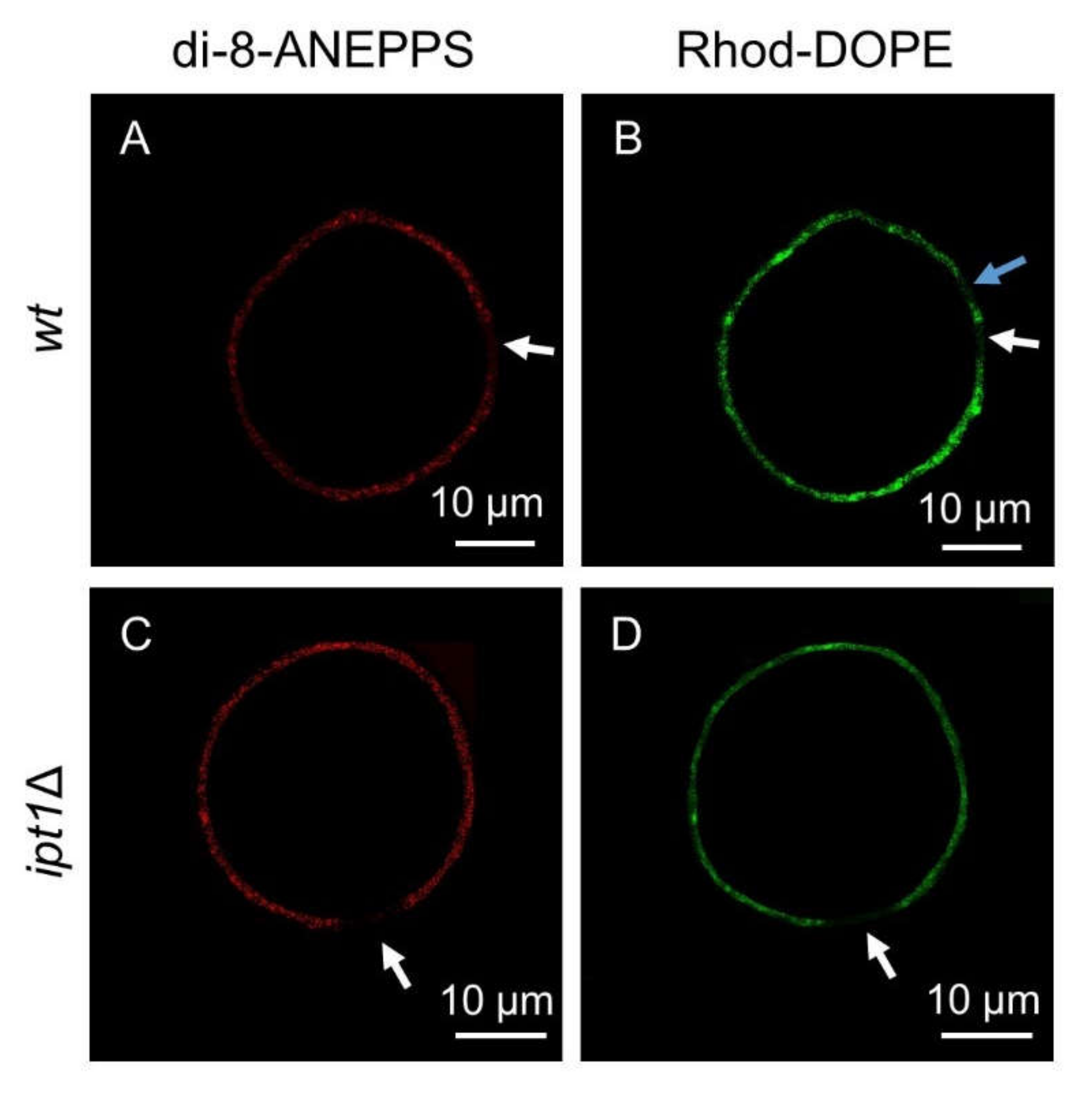
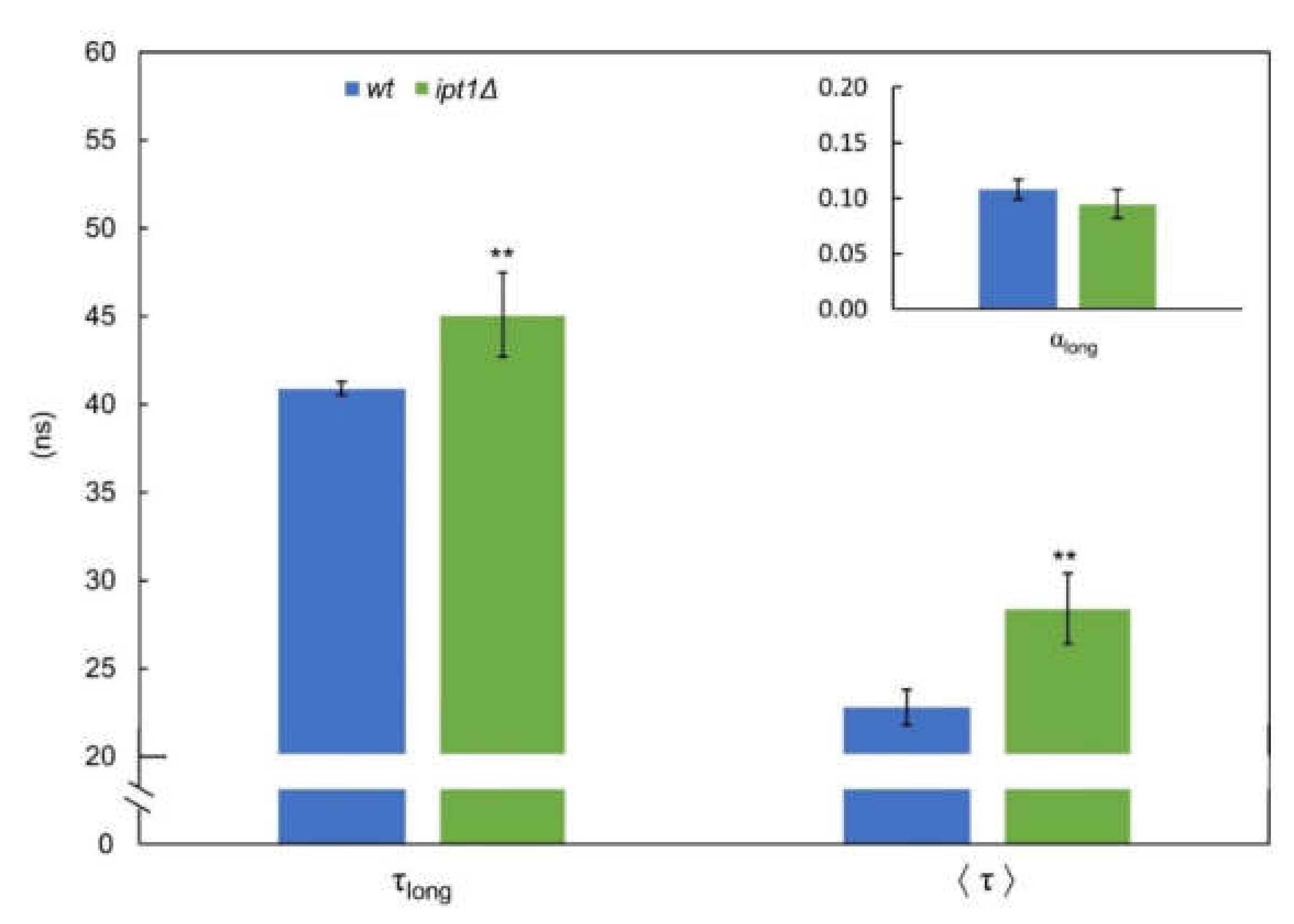
3.1. Sphingolipid-Enriched Domains are more Compact in ipt1Δ Cells
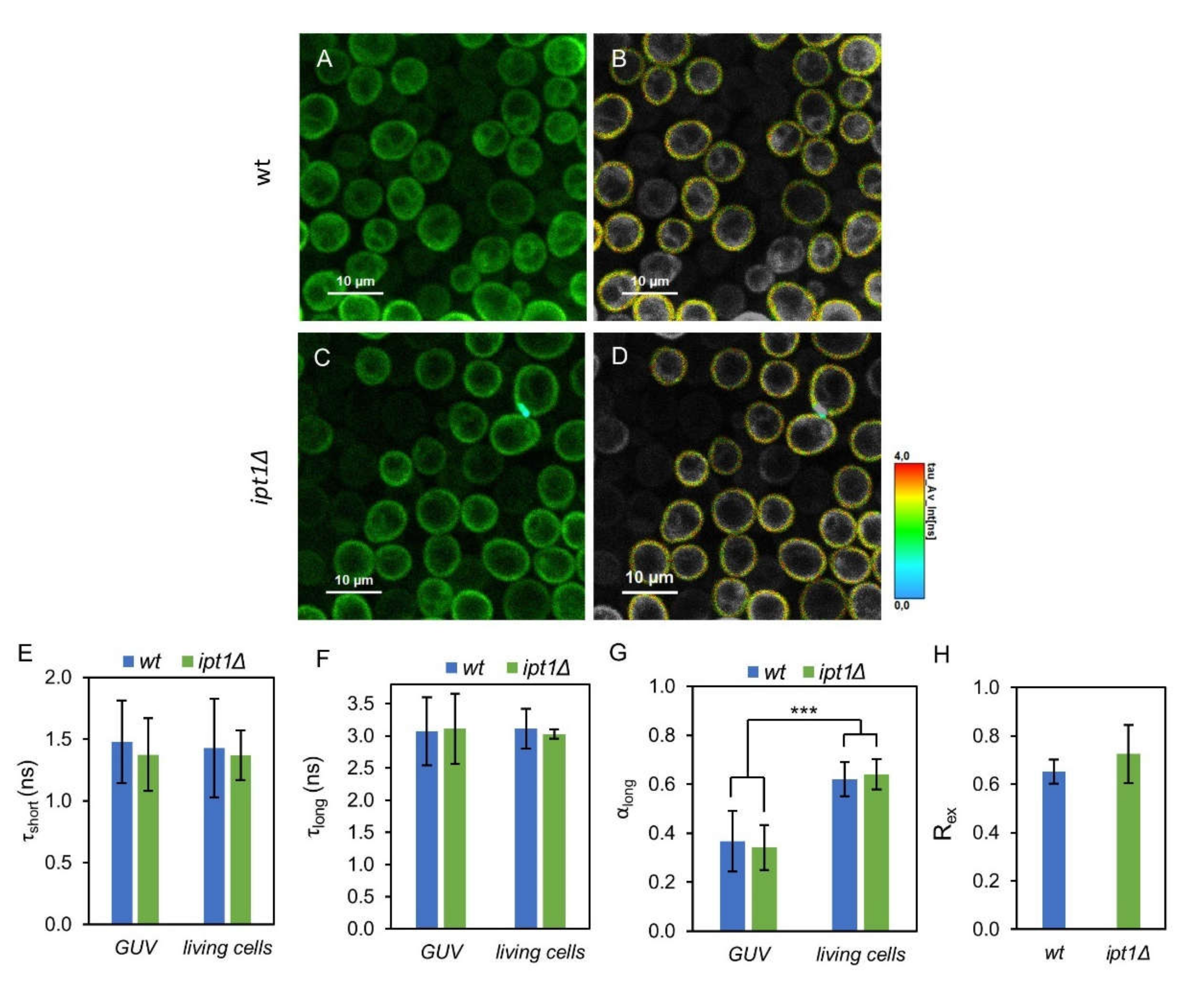
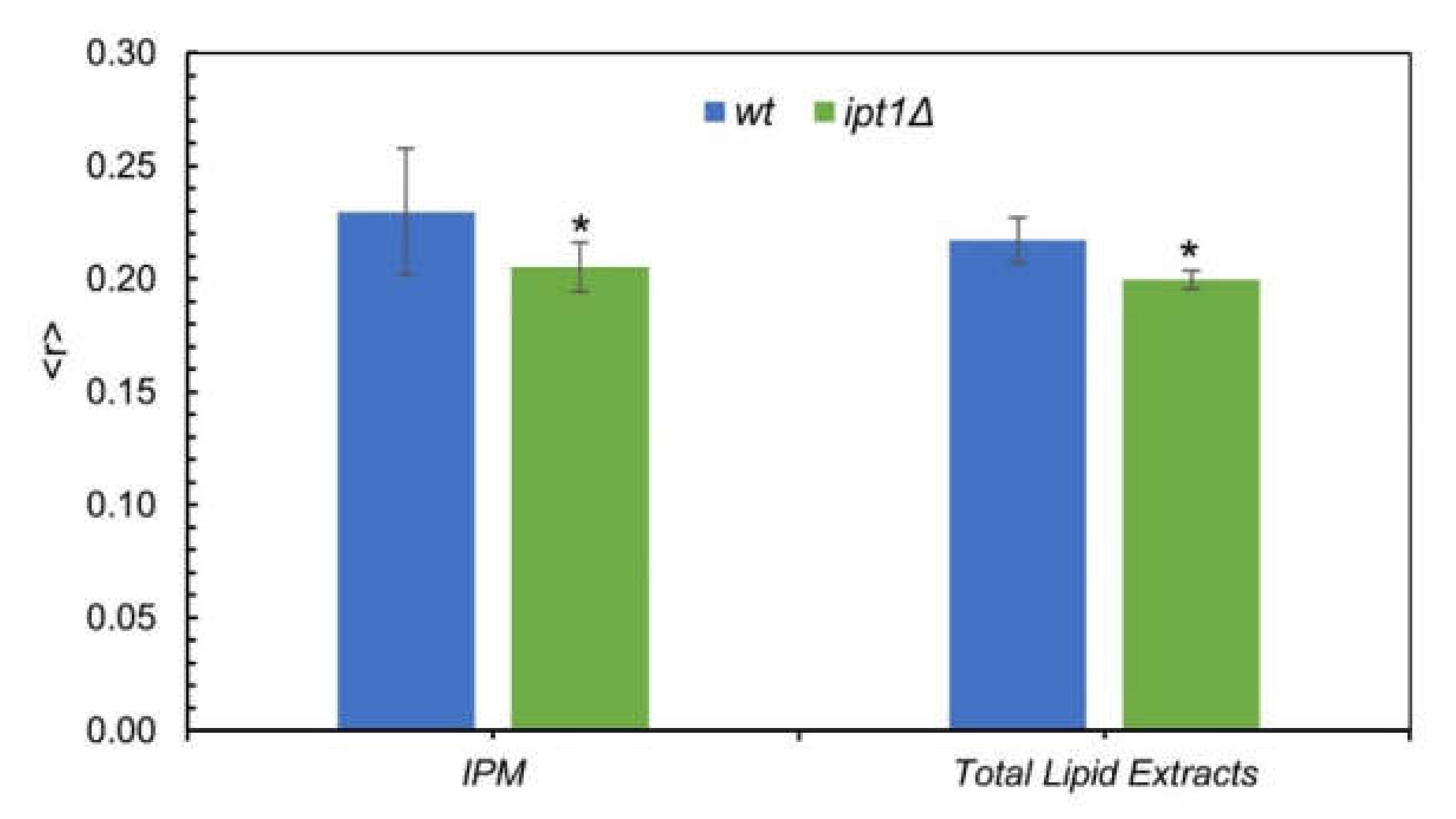
3.2. Membrane Fluidity Is Altered in ipt1Δ Cells
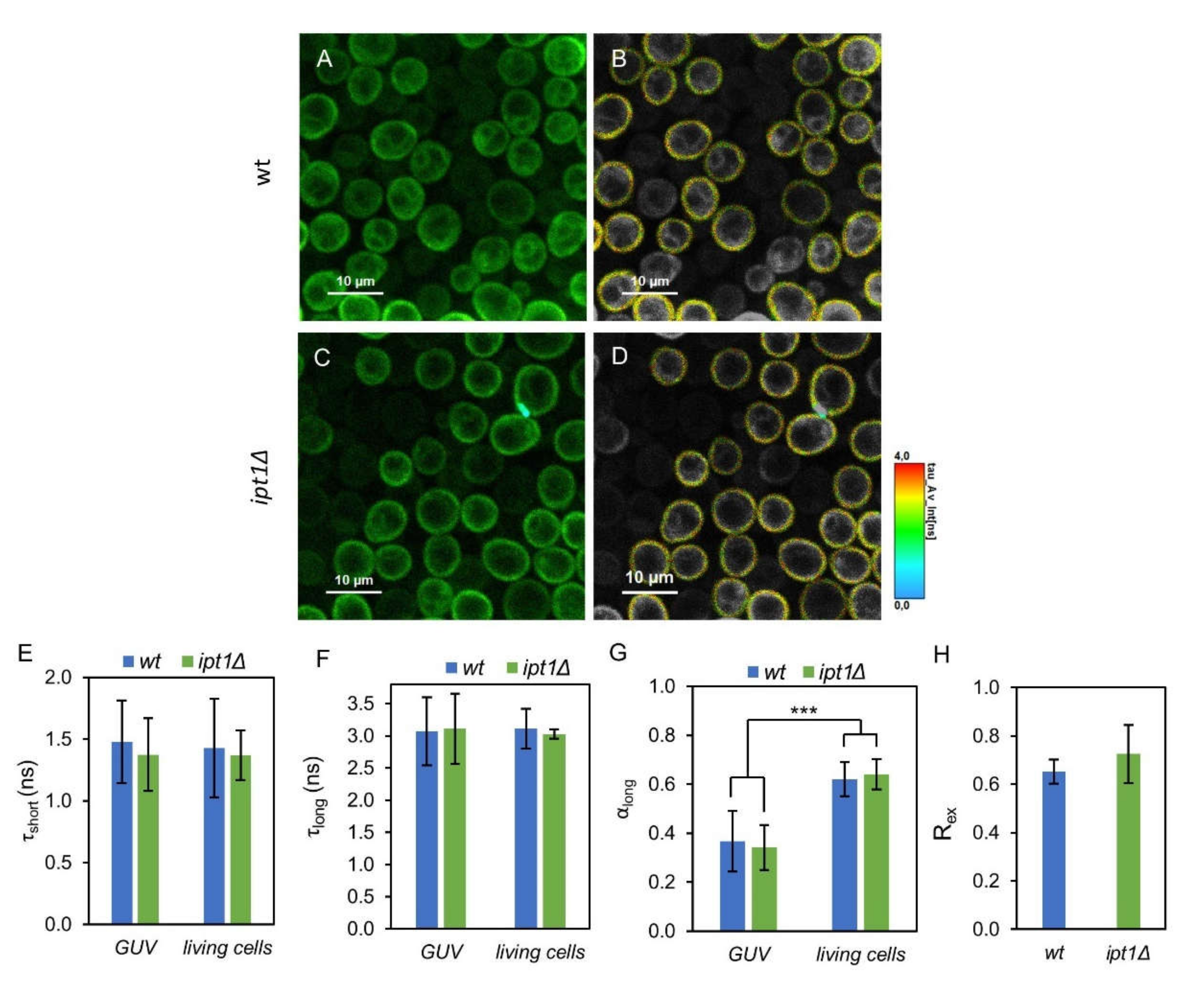
3.3. Plasma Membrane Fluid Domains Present Similar Properties in wt and ipt1Δ cells
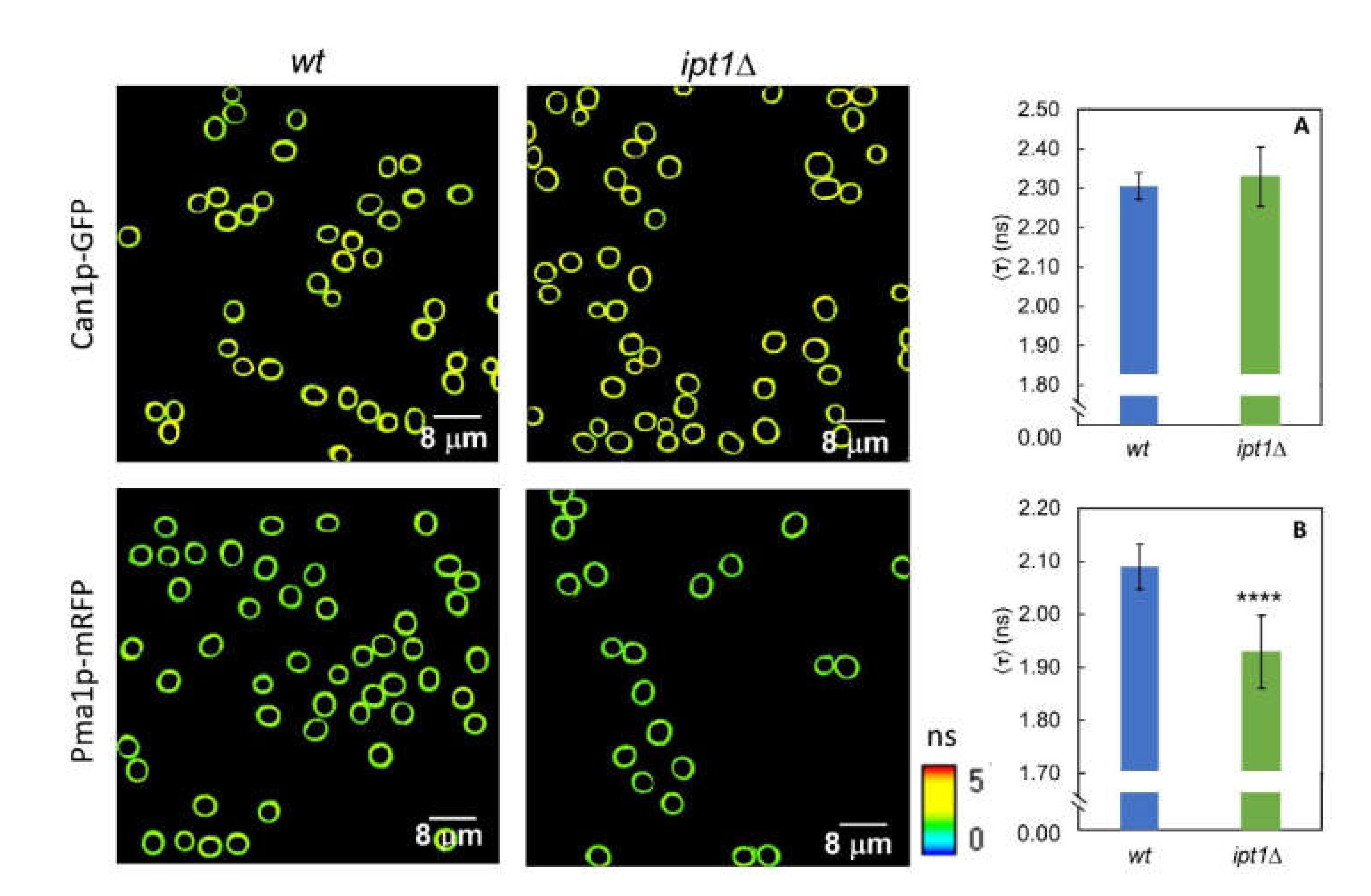
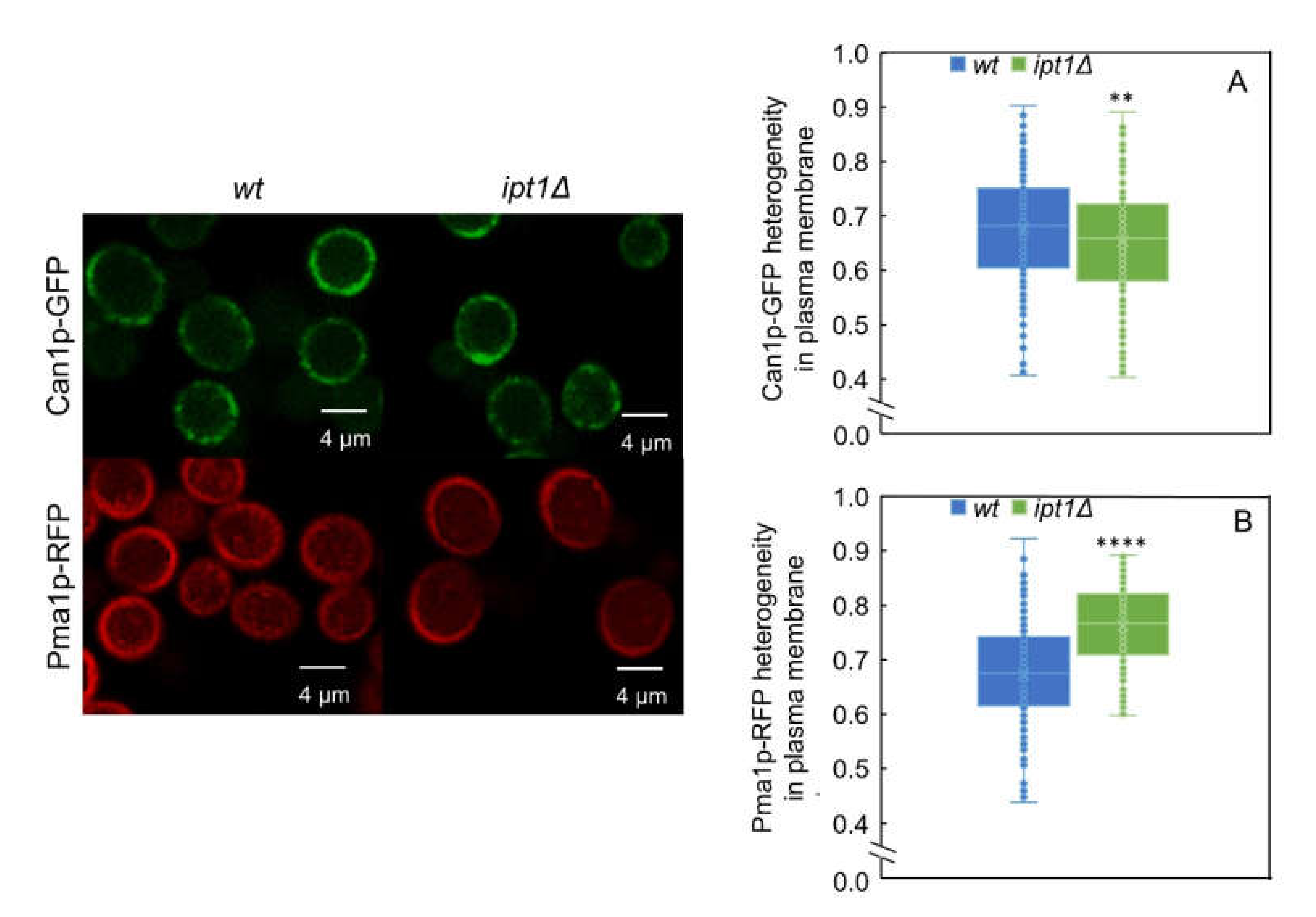
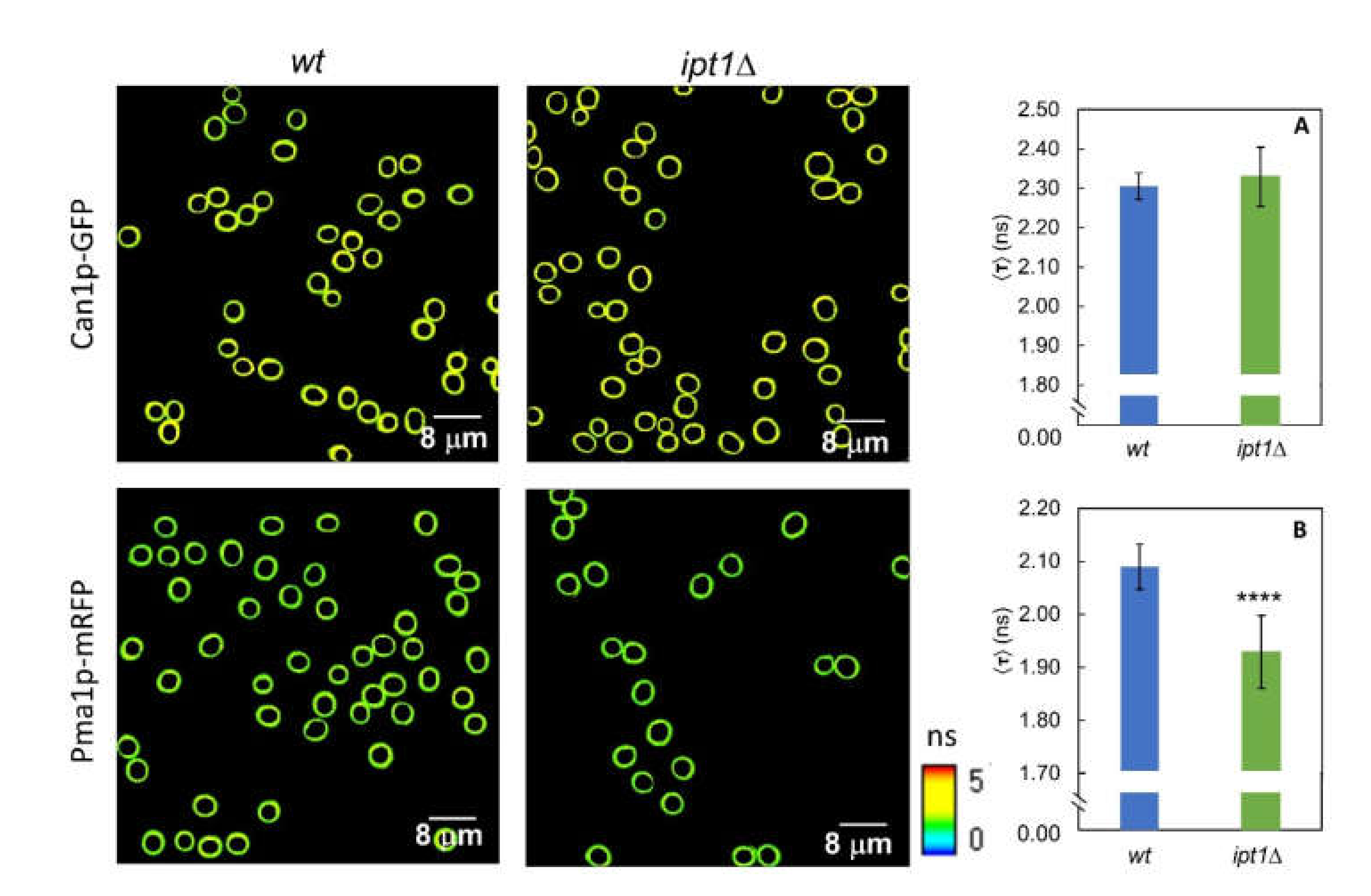
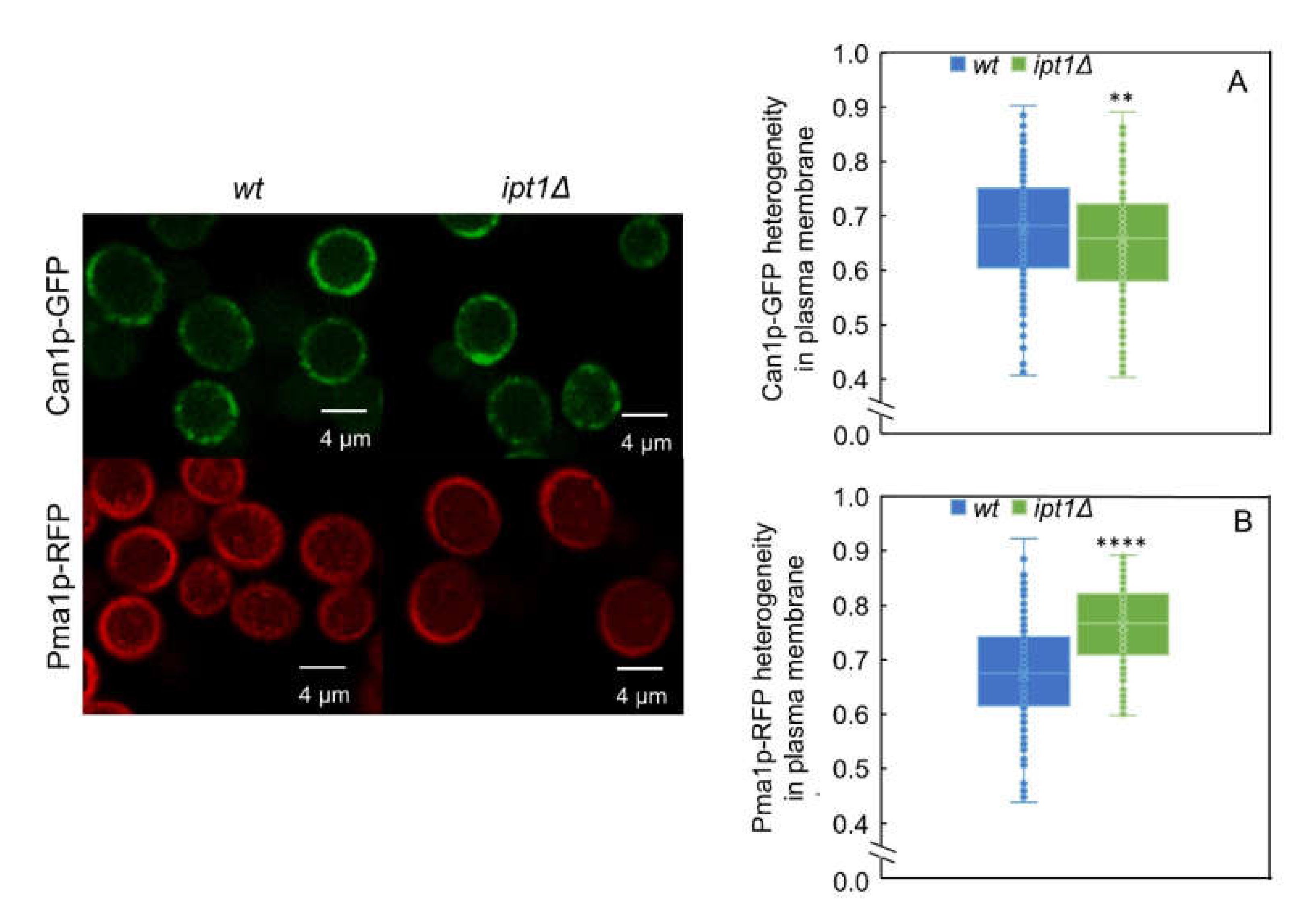
3.4. The Lateral Organization of Pma1p But Not Can1p Is Dependent on the Sphingolipid Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malinsky, J.; Opekarova, M. New Insight into the Roles of Membrane Microdomains in Physiological Activities of Fungal Cells. Int. Rev. Cell Mol. Biol. 2016, 325, 119–180. [Google Scholar] [CrossRef] [PubMed]
- Zahumensky, J.; Malinsky, J. Role of MCC/Eisosome in Fungal Lipid Homeostasis. Biomolecules 2019, 9, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasopoulos, A.; André, B.; Sophianopoulou, V.; Gournas, C. Fungal Plasma Membrane Domains. FEMS Microbiol. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, R.F.M. A route to understanding yeast cellular envelope—Plasma membrane lipids interplaying in cell wall integrity. FEBS J. 2018. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, S.; Singh, P.; Manoharlal, R.; Prasad, R.; Chattopadhyay, A. Differential dynamics of membrane proteins in yeast. Biochem. Biophys. Res. Commun. 2009, 387, 661–665. [Google Scholar] [CrossRef]
- Valdez-Taubas, J.; Pelham, H.R. Slow diffusion of proteins in the yeast plasma membrane allows polarity to be maintained by endocytic cycling. Curr. Biol. CB 2003, 13, 1636–1640. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, F.; Syga, Ł.; Moiset, G.; Spakman, D.; Schavemaker, P.E.; Punter, C.M.; Seinen, A.-B.; van Oijen, A.M.; Robinson, A.; Poolman, B. Steric exclusion and protein conformation determine the localization of plasma membrane transporters. Nat.Commun. 2018, 9, 501. [Google Scholar] [CrossRef]
- Gournas, C.; Gkionis, S.; Carquin, M.; Twyffels, L.; Tyteca, D.; André, B. Conformation-dependent partitioning of yeast nutrient transporters into starvation-protective membrane domains. Proc. Natl. Acad. Sci. USA 2018, 115, E3145–E3154. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, R.F.; Joly, E. Crystallization around solid-like nanosized docks can explain the specificity, diversity, and stability of membrane microdomains. Front. Plant Sci. 2014, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Vecer, J.; Vesela, P.; Malinsky, J.; Herman, P. Sphingolipid levels crucially modulate lateral microdomain organization of plasma membrane in living yeast. FEBS Lett. 2014, 588, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Aresta-Branco, F.; Cordeiro, A.M.; Marinho, H.S.; Cyrne, L.; Antunes, F.; de Almeida, R.F. Gel domains in the plasma membrane of Saccharomyces cerevisiae: Highly ordered, ergosterol-free, and sphingolipid-enriched lipid rafts. J. Biol. Chem. 2011, 286, 5043–5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, A.G.; Marquês, J.T.; Carreira, A.C.; Castro, I.R.; Viana, A.S.; Mingeot-Leclercq, M.P.; de Almeida, R.F.M.; Silva, L.C. The molecular mechanism of Nystatin action is dependent on the membrane biophysical properties and lipid composition. Phys. Chem. Chem. Phys. 2017, 19, 30078–30088. [Google Scholar] [CrossRef] [PubMed]
- Stradalova, V.; Stahlschmidt, W.; Grossmann, G.; Blazikova, M.; Rachel, R.; Tanner, W.; Malinsky, J. Furrow-like invaginations of the yeast plasma membrane correspond to membrane compartment of Can1. J. Cell Sci. 2009, 122, 2887–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, T.C.; Brickner, J.H.; Aguilar, P.S.; Bernales, S.; Pantoja, C.; Walter, P. Eisosomes mark static sites of endocytosis. Nature 2006, 439, 998–1003. [Google Scholar] [CrossRef]
- Grossmann, G.; Opekarova, M.; Malinsky, J.; Weig-Meckl, I.; Tanner, W. Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. EMBO J. 2007, 26, 1–8. [Google Scholar] [CrossRef]
- Berchtold, D.; Piccolis, M.; Chiaruttini, N.; Riezman, I.; Riezman, H.; Roux, A.; Walther, T.C.; Loewith, R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 2012, 14, 542–547. [Google Scholar] [CrossRef]
- Douglas, L.M.; Konopka, J.B. Plasma membrane architecture protects Candida albicans from killing by copper. PLoS Genet. 2019, 15, e1007911. [Google Scholar] [CrossRef] [Green Version]
- Douglas, L.M.; Wang, H.X.; Keppler-Ross, S.; Dean, N.; Konopka, J.B. Sur7 promotes plasma membrane organization and is needed for resistance to stressful conditions and to the invasive growth and virulence of Candida albicans. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Pedroso, N.; Matias, A.C.; Cyrne, L.; Antunes, F.; Borges, C.; Malho, R.; de Almeida, R.F.M.; Herrero, E.; Marinho, H.S. Modulation of plasma membrane lipid profile and microdomains by H2O2 in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2009, 46, 289–298. [Google Scholar] [CrossRef]
- Dupont, S.; Beney, L.; Ritt, J.F.; Lherminier, J.; Gervais, P. Lateral reorganization of plasma membrane is involved in the yeast resistance to severe dehydration. Biochim. Biophys. Acta 2010, 1798, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, G.; Malinsky, J.; Stahlschmidt, W.; Loibl, M.; Weig-Meckl, I.; Frommer, W.B.; Opekarova, M.; Tanner, W. Plasma membrane microdomains regulate turnover of transport proteins in yeast. J. Cell Biol. 2008, 183, 1075–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, P.; Vecer, J.; Opekarova, M.; Vesela, P.; Jancikova, I.; Zahumensky, J.; Malinsky, J. Depolarization affects the lateral microdomain structure of yeast plasma membrane. FEBS J. 2015, 282, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Malinska, K.; Malinsky, J.; Opekarova, M.; Tanner, W. Visualization of protein compartmentation within the plasma membrane of living yeast cells. Mol. Biol. Cell 2003, 14, 4427–4436. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R. Transport across yeast vacuolar and plasma membrane. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis, and Energetics; Broach, J.R., Jones, E.W., Pringle, J.R., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1991; Volume 21, pp. 523–585. [Google Scholar]
- Kane, P.M. Proton Transport and pH Control in Fungi. Adv. Exp. Med. Biol. 2016, 892, 33–68. [Google Scholar] [CrossRef] [Green Version]
- Bagnat, M.; Chang, A.; Simons, K. Plasma membrane proton ATPase Pma1p requires raft association for surface delivery in yeast. Mol. Biol. Cell 2001, 12, 4129–4138. [Google Scholar] [CrossRef] [Green Version]
- Gaigg, B.; Toulmay, A.; Schneiter, R. Very long-chain fatty acid-containing lipids rather than sphingolipids per se are required for raft association and stable surface transport of newly synthesized plasma membrane ATPase in yeast. J. Biol. Chem. 2006, 281, 34135–34145. [Google Scholar] [CrossRef] [Green Version]
- Farnoud, A.M.; Mor, V.; Singh, A.; Del Poeta, M. Inositol phosphosphingolipid phospholipase C1 regulates plasma membrane ATPase (Pma1) stability in Cryptococcus neoformans. FEBS Lett. 2014, 588, 3932–3938. [Google Scholar] [CrossRef] [Green Version]
- Munshi, M.A.; Gardin, J.M.; Singh, A.; Luberto, C.; Rieger, R.; Bouklas, T.; Fries, B.C.; Del Poeta, M. The Role of Ceramide Synthases in the Pathogenicity of Cryptococcus neoformans. Cell Rep. 2018, 22, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chang, A. Sphingoid base synthesis is required for oligomerization and cell surface stability of the yeast plasma membrane ATPase, Pma1. Proc. Natl. Acad. Sci. USA 2002, 99, 12853–12858. [Google Scholar] [CrossRef] [Green Version]
- Lauwers, E.; Andre, B. Association of yeast transporters with detergent-resistant membranes correlates with their cell-surface location. Traffic 2006, 7, 1045–1059. [Google Scholar] [CrossRef]
- Lauwers, E.; Grossmann, G.; Andre, B. Evidence for coupled biogenesis of yeast Gap1 permease and sphingolipids: Essential role in transport activity and normal control by ubiquitination. Mol. Biol. Cell 2007, 18, 3068–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.C.; Hamamoto, S.; Schekman, R. Ceramide biosynthesis is required for the formation of the oligomeric H+-ATPase Pma1p in the yeast endoplasmic reticulum. J. Biol. Chem. 2002, 277, 22395–22401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, T.; Mason, A.B.; Slayman, C.W. The yeast Pma1 proton pump: A model for understanding the biogenesis of plasma membrane proteins. J. Biol. Chem. 2001, 276, 29613–29616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, A.; Fischer, P.; Schneiter, R.; Kohlwein, S.D.; Daum, G. The yeast mic2 mutant is defective in the formation of mannosyl-diinositolphosphorylceramide. FEBS Lett. 1997, 411, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.C.; Nagiec, E.E.; Wells, G.B.; Nagiec, M.M.; Lester, R.L. Synthesis of mannose-(inositol-P)(2)-ceramide, the major sphingolipid in Saccharomyces cerevisiae, requires the IPT1 (YDR072c) gene. J. Biol. Chem. 1997, 272, 29620–29625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, S.; Mehlgarten, C.; Kitamoto, H.K.; Nagase, J.; Jablonowski, D.; Dickson, R.C.; Stark, M.J.R.; Schaffrath, R. Mannosyl-diinositolphospho-ceramide, the major yeast plasma membrane sphingolipid, governs toxicity of Kluyveromyces lactis zymocin. Eukaryot. Cell 2005, 4, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Stock, S.D.; Hama, H.; Radding, J.A.; Young, D.A.; Takemoto, J.Y. Syringomycin E inhibition of Saccharomyces cerevisiae: Requirement for biosynthesis of sphingolipids with very-long-chain fatty acids and mannose- and phosphoinositol-containing head groups. Antimicrob. Agents Chemother. 2000, 44, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Thevissen, K.; Cammue, B.P.; Lemaire, K.; Winderickx, J.; Dickson, R.C.; Lester, R.L.; Ferket, K.K.; Van Even, F.; Parret, A.H.; Broekaert, W.F. A gene encoding a sphingolipid biosynthesis enzyme determines the sensitivity of Saccharomyces cerevisiae to an antifungal plant defensin from dahlia (Dahlia merckii). Proc. Natl. Acad. Sci. USA 2000, 97, 9531–9536. [Google Scholar] [CrossRef] [Green Version]
- Gietz, D.; St Jean, A.; Woods, R.A.; Schiestl, R.H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992, 20, 1425. [Google Scholar] [CrossRef]
- Johnson, I.; Spence, M.T.Z. (Eds.) The Molecular Probes Handbook—A Guide to Fluorescent Probes and Labeling Technologies; Life Technologies Corporation: Carlsbad, CA, USA, 2010. [Google Scholar]
- Branco, M.R.; Marinho, H.S.; Cyrne, L.; Antunes, F. Decrease of H2O2 Plasma Membrane Permeability during Adaptation to H2O2 in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 6501–6506. [Google Scholar] [CrossRef] [Green Version]
- Panaretou, B.; Piper, P. Isolation of yeast plasma membranes. Methods Mol. Biol. 2006, 313, 27–32. [Google Scholar] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClare, C.W. An accurate and convenient organic phosphorus assay. Anal. Biochem. 1971, 39, 527–530. [Google Scholar] [CrossRef]
- Daum, G.; Tuller, G.; Nemec, T.; Hrastnik, C.; Balliano, G.; Cattel, L.; Milla, P.; Rocco, F.; Conzelmann, A.; Vionnet, C.; et al. Systematic analysis of yeast strains with possible defects in lipid metabolism. Yeast 1999, 15, 601–614. [Google Scholar] [CrossRef]
- Angelova, M.I.; Soléau, S.; Méléard, P.; Faucon, F.; Bothorel, P. Preparation of giant vesicles by external AC electric fields. Kinetics and applications. In Trends in Colloid and Interface Science VI; Steinkopff: Darmstadt, Germany, 1992; pp. 127–131. [Google Scholar]
- Marques, J.T.; Cordeiro, A.M.; Viana, A.S.; Herrmann, A.; Marinho, H.S.; de Almeida, R.F. Formation and Properties of Membrane-Ordered Domains by Phytoceramide: Role of Sphingoid Base Hydroxylation. Langmuir ACS J. Surf. Colloids 2015, 31, 9410–9421. [Google Scholar] [CrossRef]
- Bastos, A.E.; Scolari, S.; Stockl, M.; Almeida, R.F. Applications of fluorescence lifetime spectroscopy and imaging to lipid domains in vivo. In Methods Enzymol; Conn, P.M., Ed.; Academic Press: Burlington, MA, USA, 2012; Volume 504, pp. 57–81. [Google Scholar] [CrossRef]
- Haldar, S.; Kanaparthi, R.K.; Samanta, A.; Chattopadhyay, A. Differential Effect of Cholesterol and Its Biosynthetic Precursors on Membrane Dipole Potential. Biophys. J. 2012, 102, 1561–1569. [Google Scholar] [CrossRef] [Green Version]
- Khmelinskaia, A.; Marques, J.M.; Bastos, A.E.; Antunes, C.A.; Bento-Oliveira, A.; Scolari, S.; Lobo, G.M.; Herrmann, A.; Marinho, H.S.; de Almeida, R.F. Liquid-ordered phase formation by mammalian and yeast sterols: A common feature with organizational differences. Front. Cell Dev. Biol. 2020. Accepted. [Google Scholar] [CrossRef]
- Francisco, A.P.; Botequim, D.; Prazeres, D.M.F.; Serra, V.V.; Costa, S.M.B.; Laia, C.A.T.; Paulo, P.M.R. Extreme Enhancement of Single-Molecule Fluorescence from Porphyrins Induced by Gold Nanodimer Antennas. J. Phys. Chem. Lett. 2019, 10, 1542–1549. [Google Scholar] [CrossRef]
- Sklar, L.A.; Hudson, B.S.; Simoni, R.D. Conjugated polyene fatty acids as fluorescent probes: Synthetic phospholipid membrane studies. Biochemistry 1977, 16, 819–828. [Google Scholar] [CrossRef]
- De Almeida, R.F.; Loura, L.M.; Prieto, M. Membrane lipid domains and rafts: Current applications of fluorescence lifetime spectroscopy and imaging. Chem. Phys. Lipids 2009, 157, 61–77. [Google Scholar] [CrossRef]
- Mateo, C.R.; Brochon, J.C.; Lillo, M.P.; Acuna, A.U. Lipid Clustering in Bilayers Detected by the Fluorescence Kinetics and Anisotropy of Trans-Parinaric Acid. Biophys. J. 1993, 65, 2237–2247. [Google Scholar] [CrossRef] [Green Version]
- Davenport, L. [24] Fluorescence probes for studying membrane heterogeneity. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 278, pp. 487–512. [Google Scholar]
- Florine-Casteel, K.; Feigenson, G.W. On the use of partition coefficients to characterize the distribution of fluorescent membrane probes between coexisting gel and fluid lipid phases: An analysis of the partition behavior of 1,6-diphenyl-1,3,5-hexatriene. Biochim. Biophys. Acta (BBA) Biomembr. 1988, 941, 102–106. [Google Scholar] [CrossRef]
- Lentz, B.R. Membrane “Fluidity” from Fluorescent Anisotropy Measurements. In Spectroscopic Membrane Probes; Loew, M.L., Ed.; CRC press: Boca Raton, FL, USA, 1988; pp. 13–41. [Google Scholar]
- Santos, F.C.; Fernandes, A.S.; Antunes, C.A.; Moreira, F.P.; Videira, A.; Marinho, H.S.; de Almeida, R.F. Reorganization of plasma membrane lipid domains during conidial germination. Biochim. Biophys. Acta 2017, 1862, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.; Coutinho, A.; Fedorov, A.; Prieto, M. Competitive binding of cholesterol and ergosterol to the polyene antibiotic nystatin. A fluorescence study. Biophys. J. 2006, 90, 3625–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaro, M.; Reina, F.; Hof, M.; Eggeling, C.; Sezgin, E. Laurdan and Di-4-ANEPPDHQ probe different properties of the membrane. J. Phys. D Appl. Phys. 2017, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, A.E.P.; Marinho, H.S.; Cordeiro, A.M.; de Soure, A.M.; de Almeida, R.F.M. Biophysical properties of ergosterol-enriched lipid rafts in yeast and tools for their study: Characterization of ergosterol/phosphatidylcholine membranes with three fluorescent membrane probes. Chem. Phys. Lipids 2012, 165, 577–588. [Google Scholar] [CrossRef]
- De Almeida, R.F.; Borst, J.; Fedorov, A.; Prieto, M.; Visser, A.J. Complexity of lipid domains and rafts in giant unilamellar vesicles revealed by combining imaging and microscopic and macroscopic time-resolved fluorescence. Biophys. J. 2007, 93, 539–553. [Google Scholar] [CrossRef] [Green Version]
- González-Ramírez, E.J.; Goñi, F.M.; Alonso, A. Mixing brain cerebrosides with brain ceramides, cholesterol and phospholipids. Sci. Rep. 2019, 9, 13326. [Google Scholar] [CrossRef] [Green Version]
- Pinto, S.N.; Fernandes, F.; Fedorov, A.; Futerman, A.H.; Silva, L.C.; Prieto, M. A combined fluorescence spectroscopy, confocal and 2-photon microscopy approach to re-evaluate the properties of sphingolipid domains. Biochim. Biophys. Acta 2013, 1828, 2099–2110. [Google Scholar] [CrossRef] [Green Version]
- Fluhler, E.; Burnham, V.G.; Loew, L.M. Spectra, membrane binding, and potentiometric responses of new charge shift probes. Biochemistry 1985, 24, 5749–5755. [Google Scholar] [CrossRef]
- Cotlet, M.; Hofkens, J.; Maus, M.; Gensch, T.; Van der Auweraer, M.; Michiels, J.; Dirix, G.; Van Guyse, M.; Vanderleyden, J.; Visser, A.J.W.G.; et al. Excited-State Dynamics in the Enhanced Green Fluorescent Protein Mutant Probed by Picosecond Time-Resolved Single Photon Counting Spectroscopy. J. Phys. Chem. B 2001, 105, 4999–5006. [Google Scholar] [CrossRef] [Green Version]
- Heikal, A.A.; Hess, S.T.; Webb, W.W. Multiphoton molecular spectroscopy and excited-state dynamics of enhanced green fluorescent protein (EGFP): Acid–base specificity. Chem. Phys. 2001, 274, 37–55. [Google Scholar] [CrossRef]
- Suhling, K.; Siegel, J.; Phillips, D.; French, P.M.W.; Lévêque-Fort, S.; Webb, S.E.D.; Davis, D.M. Imaging the Environment of Green Fluorescent Protein. Biophys. J. 2002, 83, 3589–3595. [Google Scholar] [CrossRef]
- Suhling, K.; Davis, D.; Petrasek, Z.; Siegel, J.; Phillips, D. Influence of the refractive index on EGFP fluorescence lifetimes in mixtures of water and glycerol. Proc. SPIE 2001, 4259, 92–101. [Google Scholar] [CrossRef]
- Petrov, J.G.; Pfohl, T.; Möhwald, H. Ellipsometric Chain Length Dependence of Fatty Acid Langmuir Monolayers. A Heads-and-Tails Model. J. Phys. Chem. B 1999, 103, 3417–3424. [Google Scholar] [CrossRef]
- Kienle, D.F.; de Souza, J.V.; Watkins, E.B.; Kuhl, T.L. Thickness and refractive index of DPPC and DPPE monolayers by multiple-beam interferometry. Anal. Bioanal. Chem. 2014, 406, 4725–4733. [Google Scholar] [CrossRef]
- Fajardo-Somera, R.A.; Bowman, B.; Riquelme, M. The plasma membrane proton pump PMA-1 is incorporated into distal parts of the hyphae independently of the Spitzenkorper in Neurospora crassa. Eukaryot. Cell 2013, 12, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Clay, L.; Caudron, F.; Denoth-Lippuner, A.; Boettcher, B.; Frei, S.B.; Snapp, E.L.; Barral, Y. A sphingolipid-dependent diffusion barrier confines ER stress to the yeast mother cell. Elife 2014, 3. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef] [Green Version]
- Gaigg, B.; Timischl, B.; Corbino, L.; Schneiter, R. Synthesis of sphingolipids with very long chain fatty acids but not ergosterol is required for routing of newly synthesized plasma membrane ATPase to the cell surface of yeast. J. Biol. Chem. 2005, 280, 22515–22522. [Google Scholar] [CrossRef] [Green Version]
- Marques, J.T.; Marinho, H.S.; de Almeida, R.F.M. Sphingolipid hydroxylation in mammals, yeast and plants—An integrated view. Prog. Lipid Res. 2018, 71, 18–42. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Toume, M. Alteration of complex sphingolipid composition and its physiological significance in yeast Saccharomyces cerevisiae lacking vacuolar ATPase. Microbiology 2015, 161, 2369–2383. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.C. Thematic Review Series: Sphingolipids. New insights into sphingolipid metabolism and function in budding yeast. J. Lipid Res. 2008, 49, 909–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallstrom, T.C.; Lambert, L.; Schorling, S.; Balzi, E.; Goffeau, A.; Moye-Rowley, W.S. Coordinate control of sphingolipid biosynthesis and multidrug resistance in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 23674–23680. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Tani, M. Mannosylinositol phosphorylceramides and ergosterol coodinately maintain cell wall integrity in the yeast Saccharomyces cerevisiae. FEBS J. 2018, 285, 2405–2427. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, Y.; Tani, M. Synthesis of mannosylinositol phosphorylceramides is involved in maintenance of cell integrity of yeast Saccharomyces cerevisiae. Mol. Microbiol. 2015, 95, 706–722. [Google Scholar] [CrossRef]
- Kean, L.S.; Grant, A.M.; Angeletti, C.; Mahé, Y.; Kuchler, K.; Fuller, R.S.; Nichols, J.W. Plasma membrane translocation of fluorescent-labeled phosphatidylethanolamine is controlled by transcription regulators, PDR1 and PDR3. J. Cell Biol. 1997, 138, 255–270. [Google Scholar] [CrossRef] [Green Version]
- Decottignies, A.; Grant, A.M.; Nichols, J.W.; de Wet, H.; McIntosh, D.B.; Goffeau, A. ATPase and Multidrug Transport Activities of the Overexpressed Yeast ABC Protein Yor1p. J. Biol. Chem. 1998, 273, 12612–12622. [Google Scholar] [CrossRef] [Green Version]
- Ratto, T.V.; Longo, M.L. Obstructed diffusion in phase-separated supported lipid bilayers: A combined atomic force microscopy and fluorescence recovery after photobleaching approach. Biophys. J. 2002, 83, 3380–3392. [Google Scholar] [CrossRef] [Green Version]
- Van‘t Klooster, J.S.; Cheng, T.Y.; Sikkema, H.R.; Jeucken, A.; Moody, B.; Poolman, B. Periprotein lipidomes of Saccharomyces cerevisiae provide a flexible environment for conformational changes of membrane proteins. Elife 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.L.; Souza, C.M.; Pichler, H.; Dewhurst, G.; Schaad, O.; Kajiwara, K.; Wakabayashi, H.; Ivanova, T.; Castillon, G.A.; Piccolis, M.; et al. Functional Interactions between Sphingolipids and Sterols in Biological Membranes Regulating Cell Physiology. Mol. Biol. Cell 2009, 20, 2083–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, E.; Baudry, K.; Stukey, J.; McDonough, V.; Germann, M.; Nickels, J.T. Sterol-dependent regulation of sphingolipid metabolism in Saccharomyces cerevisiae. J. Biol. Chem. 2002, 277, 26177–26184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanko, L.M.; Sullivan, D.P.; Sere, Y.Y.; Szomek, M.; Lunding, A.; Solanko, K.A.; Pizovic, A.; Stanchev, L.D.; Pomorski, T.G.; Menon, A.K.; et al. Ergosterol is mainly located in the cytoplasmic leaflet of the yeast plasma membrane. Traffic 2018, 19, 198–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, B.M.; de Almeida, R.F.M.; Fedorov, A.; Prieto, M. The photophysics of a Rhodamine head labeled phospholipid in the identification and characterization of membrane lipid phases. Chem. Phys. Lipids 2012, 165, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; Gow, N.A.; Bowen, D.V. Cytoplasmic alkalinization during germ tube formation in Candida albicans. J. Gen. Microbiol. 1988, 134, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahanty, S.K.; Gupta, P.; Banerjee, U.; Fotedar, R.; Prasad, R. Defective plasma membrane H(+)-ATPase or orthovanadate resistant mutants from Candida albicans, a pathogenic yeast. Biochem. Int. 1990, 22, 11–20. [Google Scholar]
- Stock, S.D.; Hama, H.; DeWald, D.B.; Takemoto, J.Y. SEC14-dependent secretion in Saccharomyces cerevisiae. Nondependence on sphingolipid synthesis-coupled diacylglycerol production. J. Biol. Chem. 1999, 274, 12979–12983. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Source | Growth Media |
|---|---|---|---|
| wild-type (wt; BY4741) | MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0 | EUROSCARF (Frankfurt, Germany) | Synthetic Complete medium (SC) |
| ipt1Δ | BY4741; YMR272c::kanMX4 | EUROSCARF (Frankfurt, Germany) | SC |
| wt-Can1p-GFP | BY4741; Ylp211CAN::GFP | This study | SC ura- |
| wt-Pma1p-mRFP | BY4741; Ylp128PMA1:mRFP | This study | SC leu- |
| ipt1Δ-Can1p-GFP | BY4741; YMR272c::kanMX4; Ylp211CAN::GFP | This study | SC ura- |
| ipt1Δ-Pma1p-mRFP | BY4741; YMR272c::kanMX4; Ylp128PMA1::mRFP | This study | SC leu- |
| Membrane System | Strain | τ1 (ns) | α1 | τ2 (ns) | α2 | (ns) | 〈τ〉 (ns) |
|---|---|---|---|---|---|---|---|
| GUVs | wt | 1.48 ± 0.33 | 0.63 ± 0.12 | 3.07 ± 0.53 | 0.37 ± 0.12 | 2.05 ± 0.32 | 2.35 ± 0.35 |
| ipt1Δ | 1.37 ± 0.29 | 0.66 ± 0.09 | 3.11 ± 0.54 | 0.34 ± 0.09 | 1.95 ± 0.33 | 2.30 ± 0.37 | |
| Living cells | wt | 1.43 ± 0.40 | 0.38 ± 0.07 | 3.00 ± 0.31 | 0.62 ± 0.07 | 2.43 ± 0.12 | 2.70 ± 0.06 |
| ipt1Δ | 1.37 ± 0.20 | 0.36 ± 0.06 | 3.03 ± 0.07 | 0.64 ± 0.06 | 2.43 ± 0.15 | 2.69 ± 0.07 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bento-Oliveira, A.; Santos, F.C.; Marquês, J.T.; Paulo, P.M.R.; Korte, T.; Herrmann, A.; Marinho, H.S.; de Almeida, R.F.M. Yeast Sphingolipid-Enriched Domains and Membrane Compartments in the Absence of Mannosyldiinositolphosphorylceramide. Biomolecules 2020, 10, 871. https://doi.org/10.3390/biom10060871
Bento-Oliveira A, Santos FC, Marquês JT, Paulo PMR, Korte T, Herrmann A, Marinho HS, de Almeida RFM. Yeast Sphingolipid-Enriched Domains and Membrane Compartments in the Absence of Mannosyldiinositolphosphorylceramide. Biomolecules. 2020; 10(6):871. https://doi.org/10.3390/biom10060871
Chicago/Turabian StyleBento-Oliveira, Andreia, Filipa C. Santos, Joaquim Trigo Marquês, Pedro M. R. Paulo, Thomas Korte, Andreas Herrmann, H. Susana Marinho, and Rodrigo F. M. de Almeida. 2020. "Yeast Sphingolipid-Enriched Domains and Membrane Compartments in the Absence of Mannosyldiinositolphosphorylceramide" Biomolecules 10, no. 6: 871. https://doi.org/10.3390/biom10060871
APA StyleBento-Oliveira, A., Santos, F. C., Marquês, J. T., Paulo, P. M. R., Korte, T., Herrmann, A., Marinho, H. S., & de Almeida, R. F. M. (2020). Yeast Sphingolipid-Enriched Domains and Membrane Compartments in the Absence of Mannosyldiinositolphosphorylceramide. Biomolecules, 10(6), 871. https://doi.org/10.3390/biom10060871