Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression


 ,
,  , ,
, ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Antibodies, and Plasmids
2.2. Cell Culture
2.3. Real-Time RT-PCR Assays
2.4. Immunofluorescence
2.5. Immunoblot Analysis
2.6. [3H]Thymidine Incorporation
2.7. Wound Healing Assays
2.8. Transmigration Assays
2.9. Neurosphere Culture
2.10. Flow Cytometry
2.11. Soft Agar Assay
2.12. Limiting Dilution Assay (LDA)
2.13. Transient Transfection Assays
2.14. Statistical Analysis
3. Results
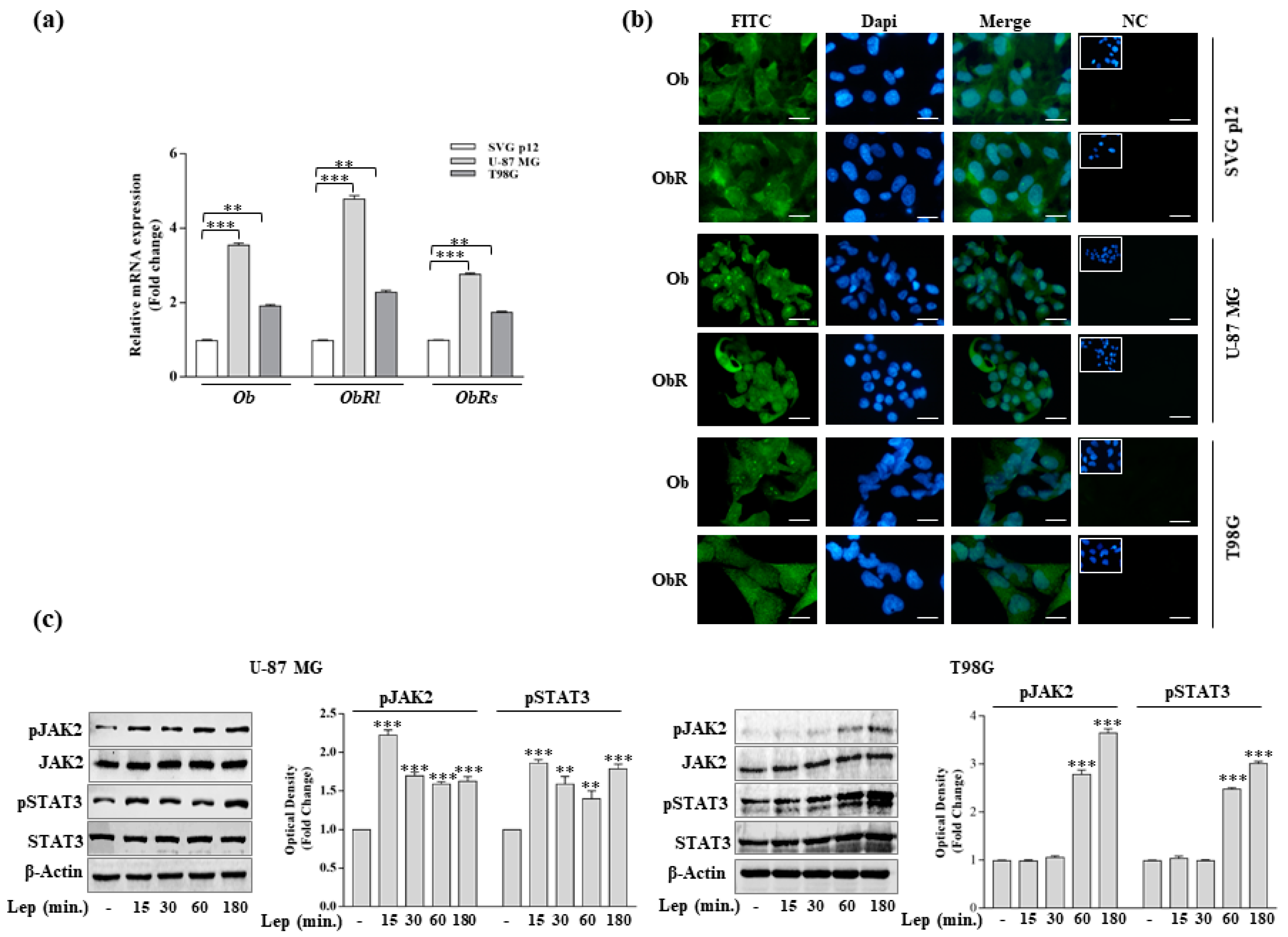
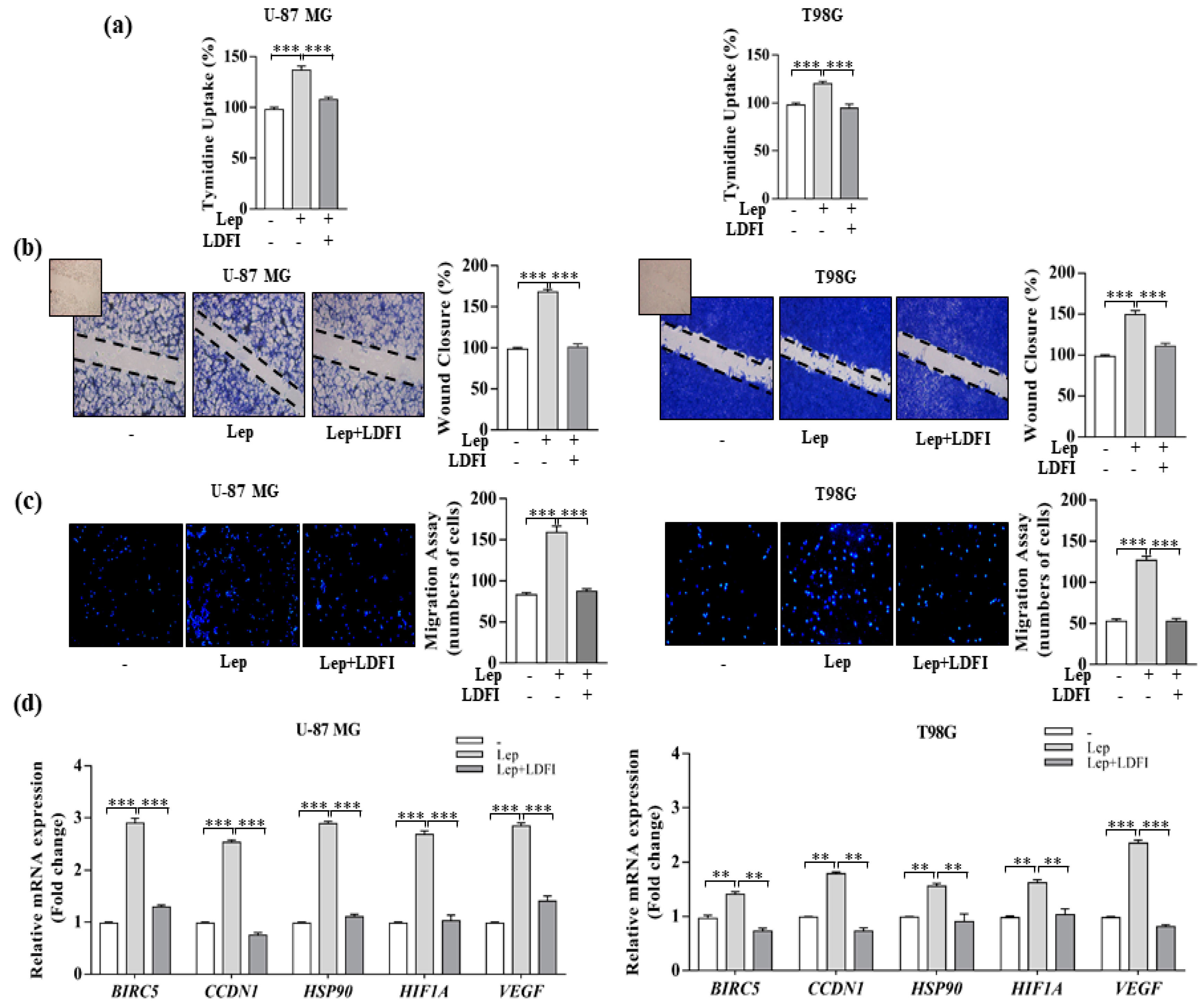
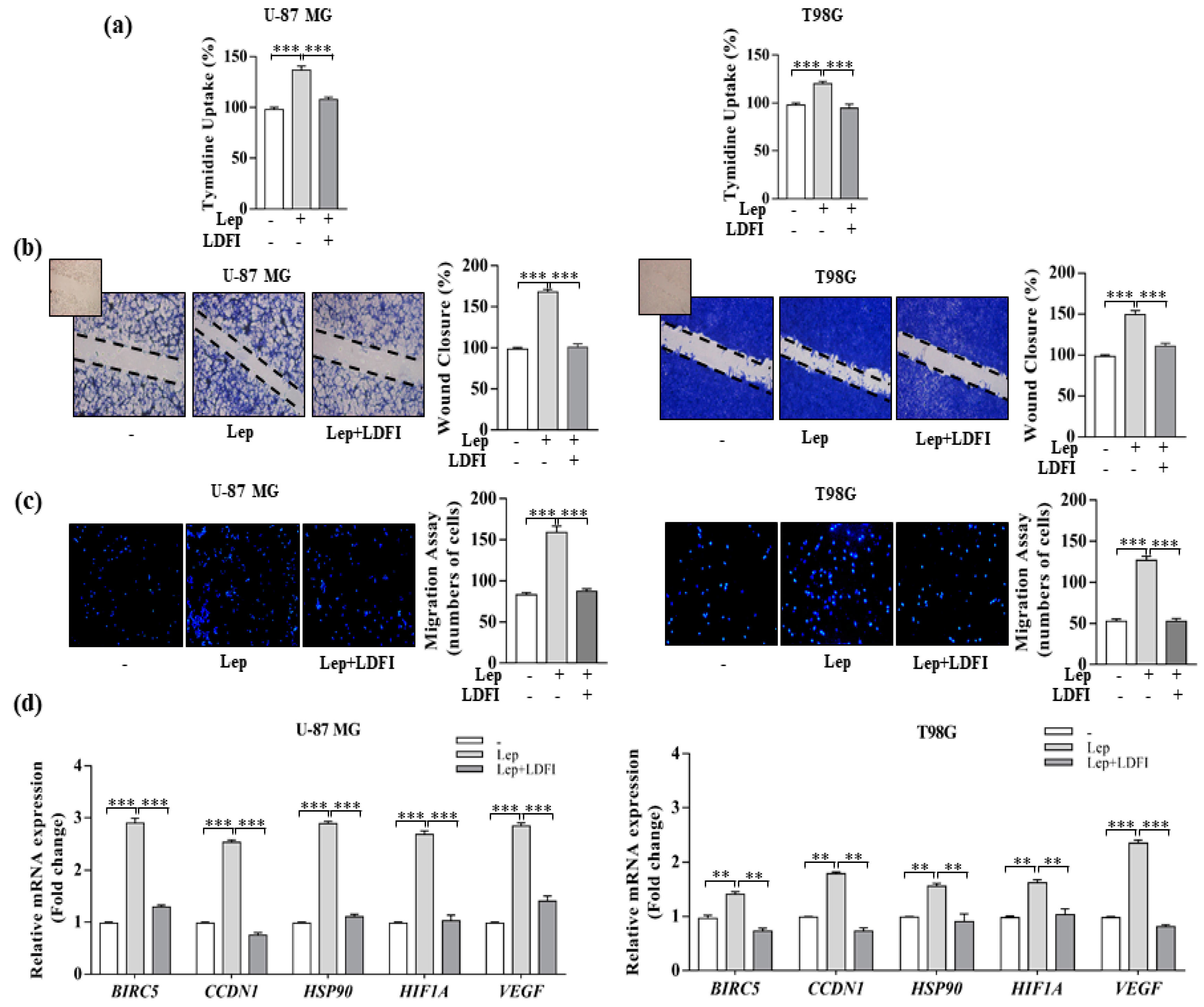
3.1. Leptin Induces Growth and Migration of GBM Cell Lines
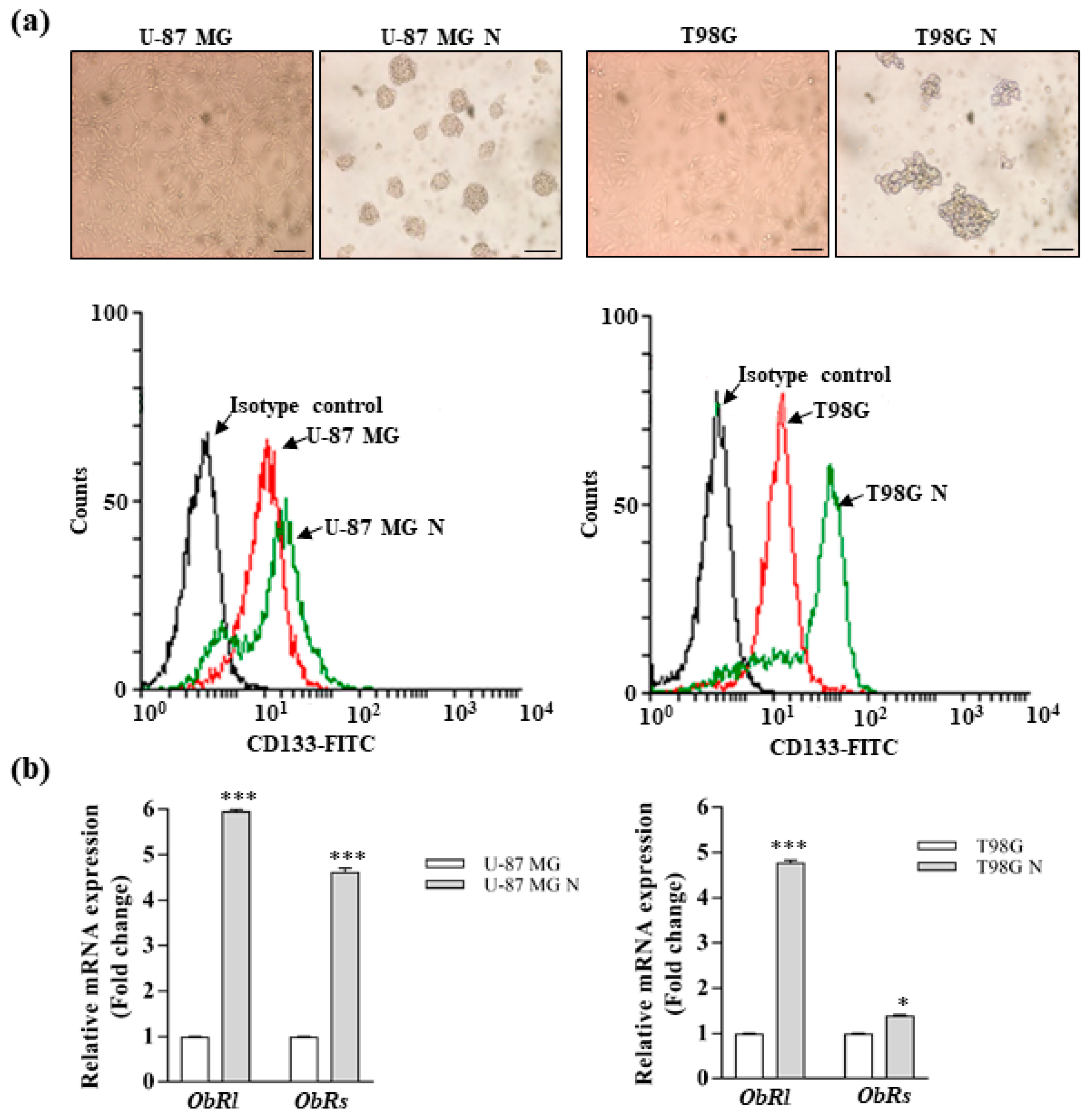
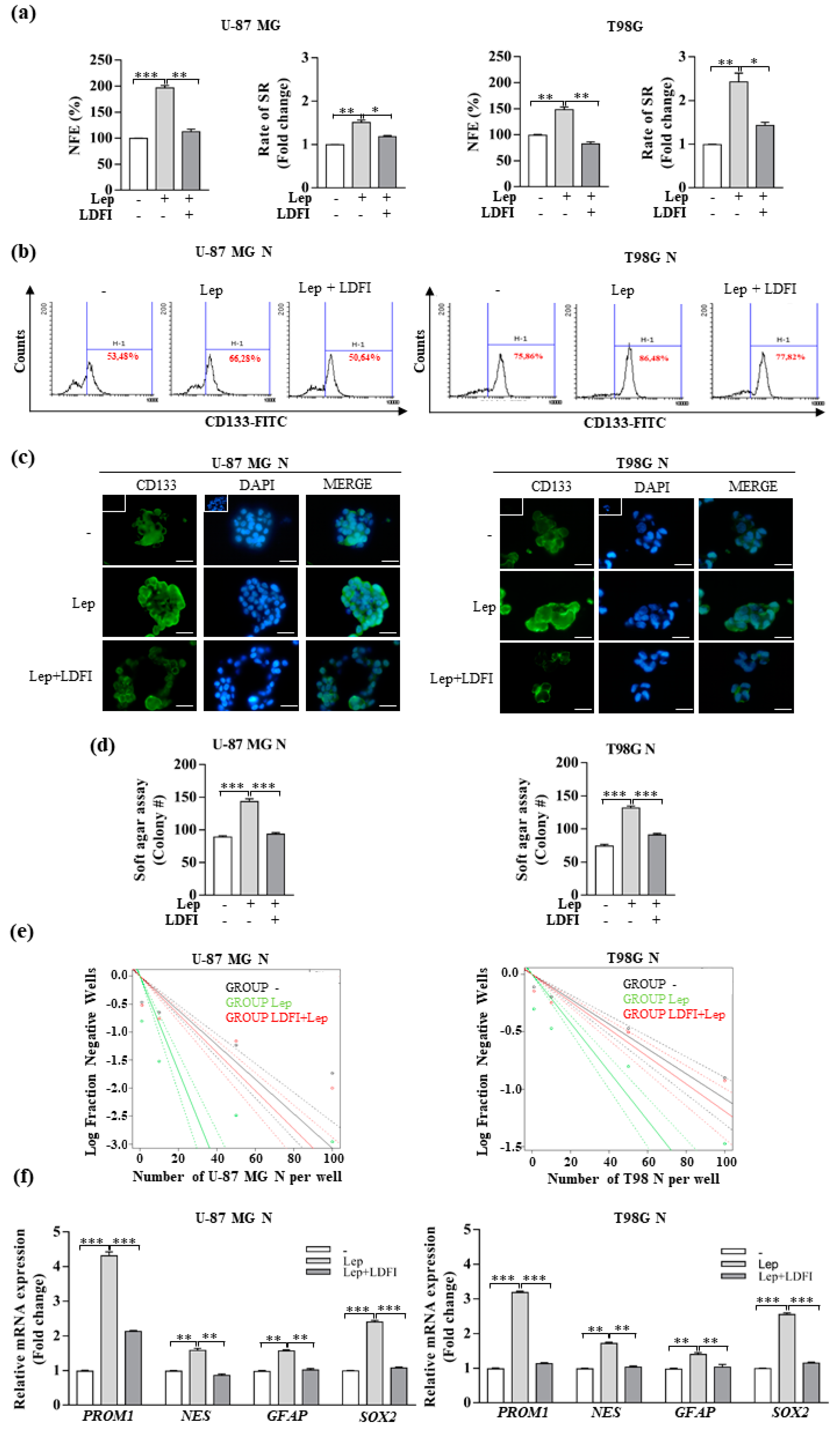
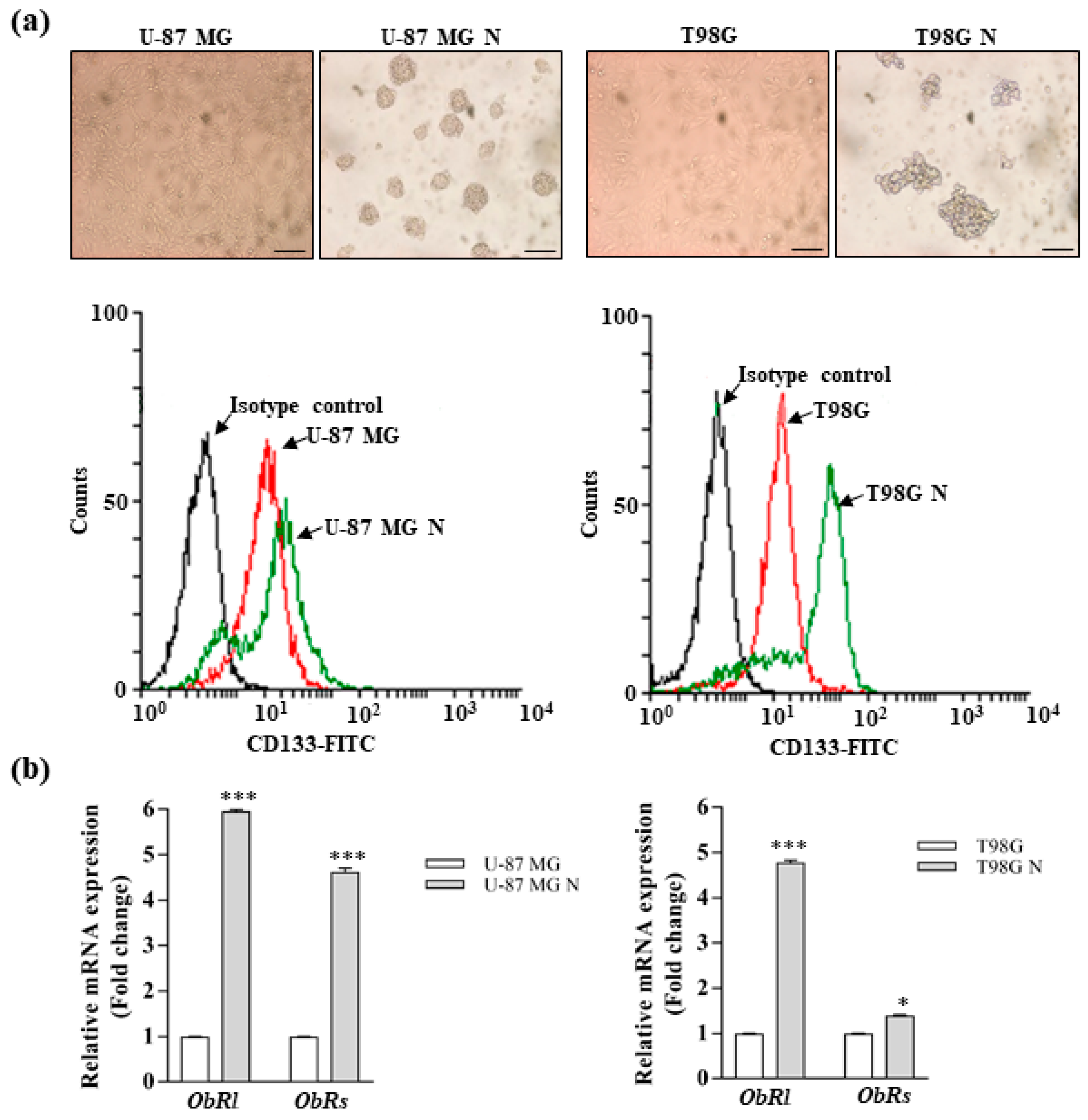
3.2. Leptin Promotes the Stemness of Glioblastoma Cells
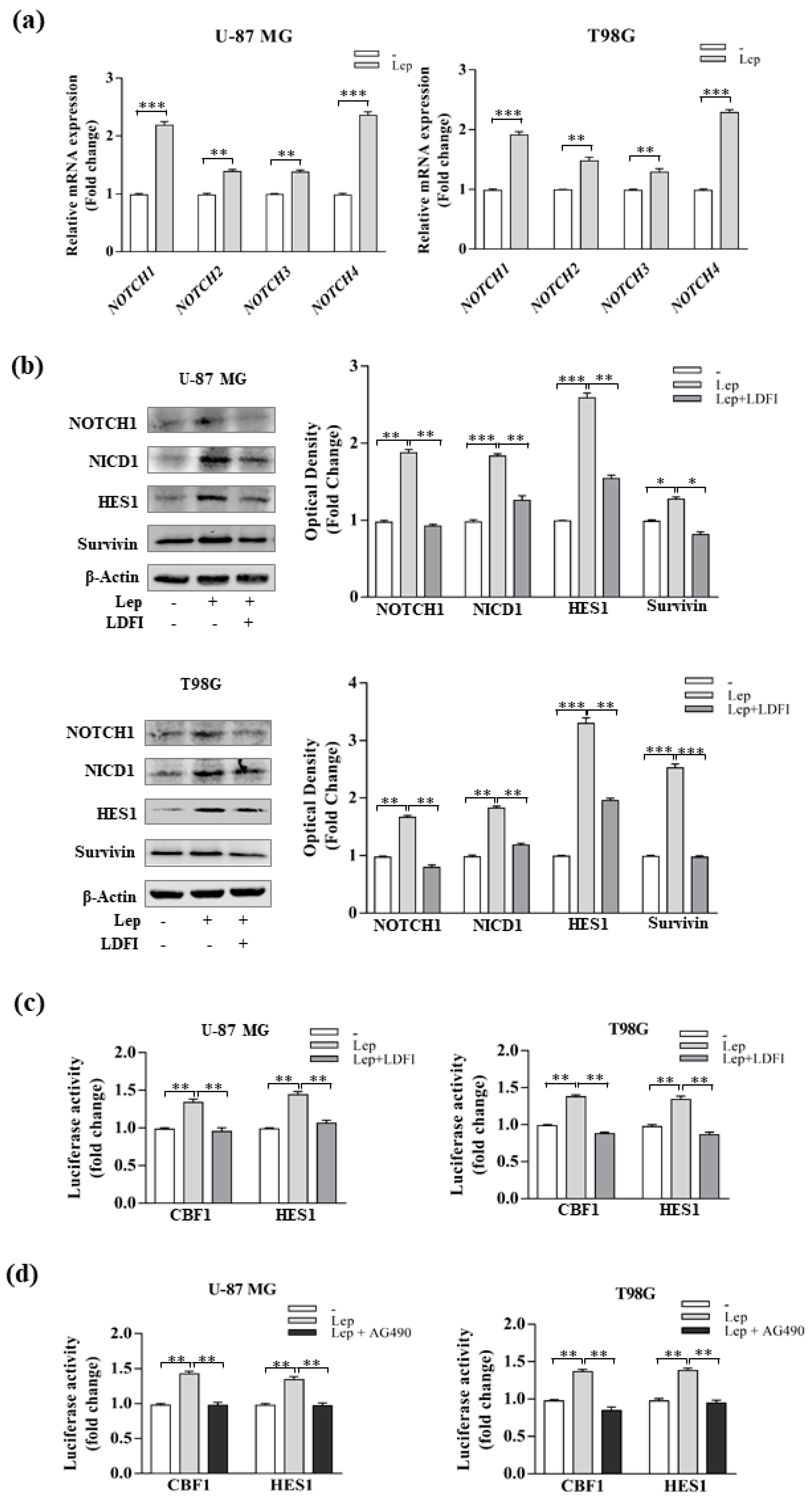
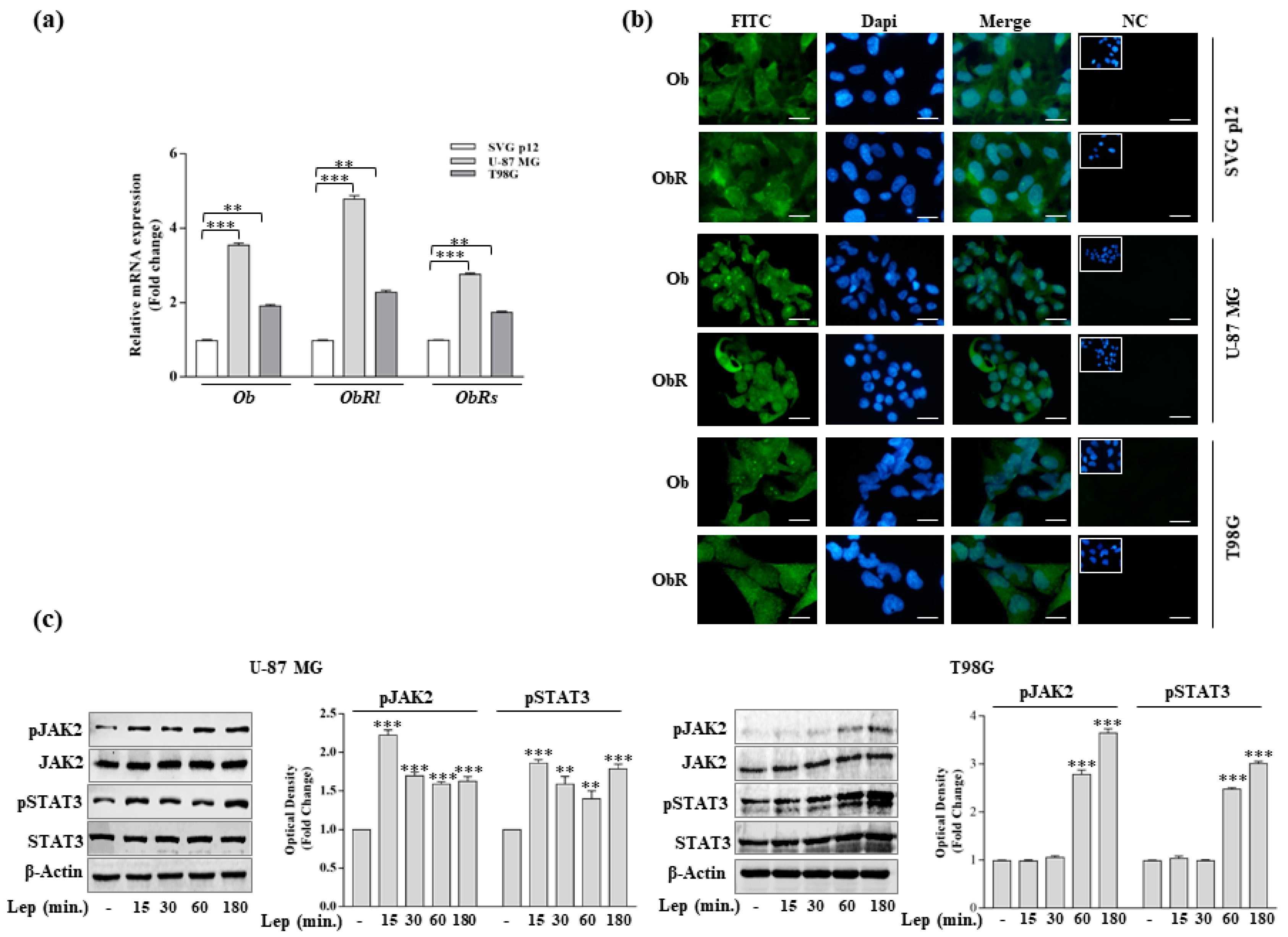
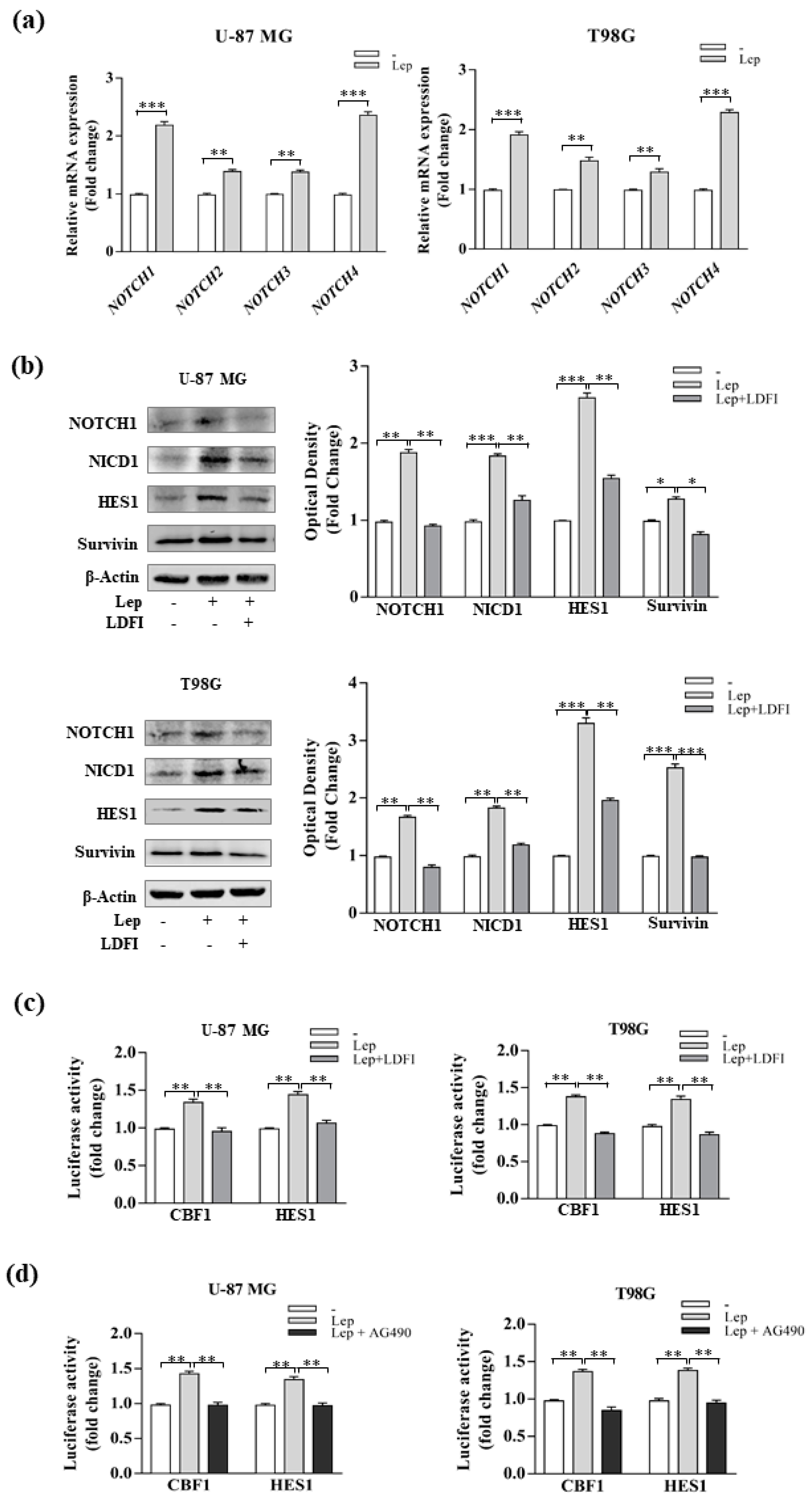
3.3. Leptin Induces Notch Expression in GBM Cells
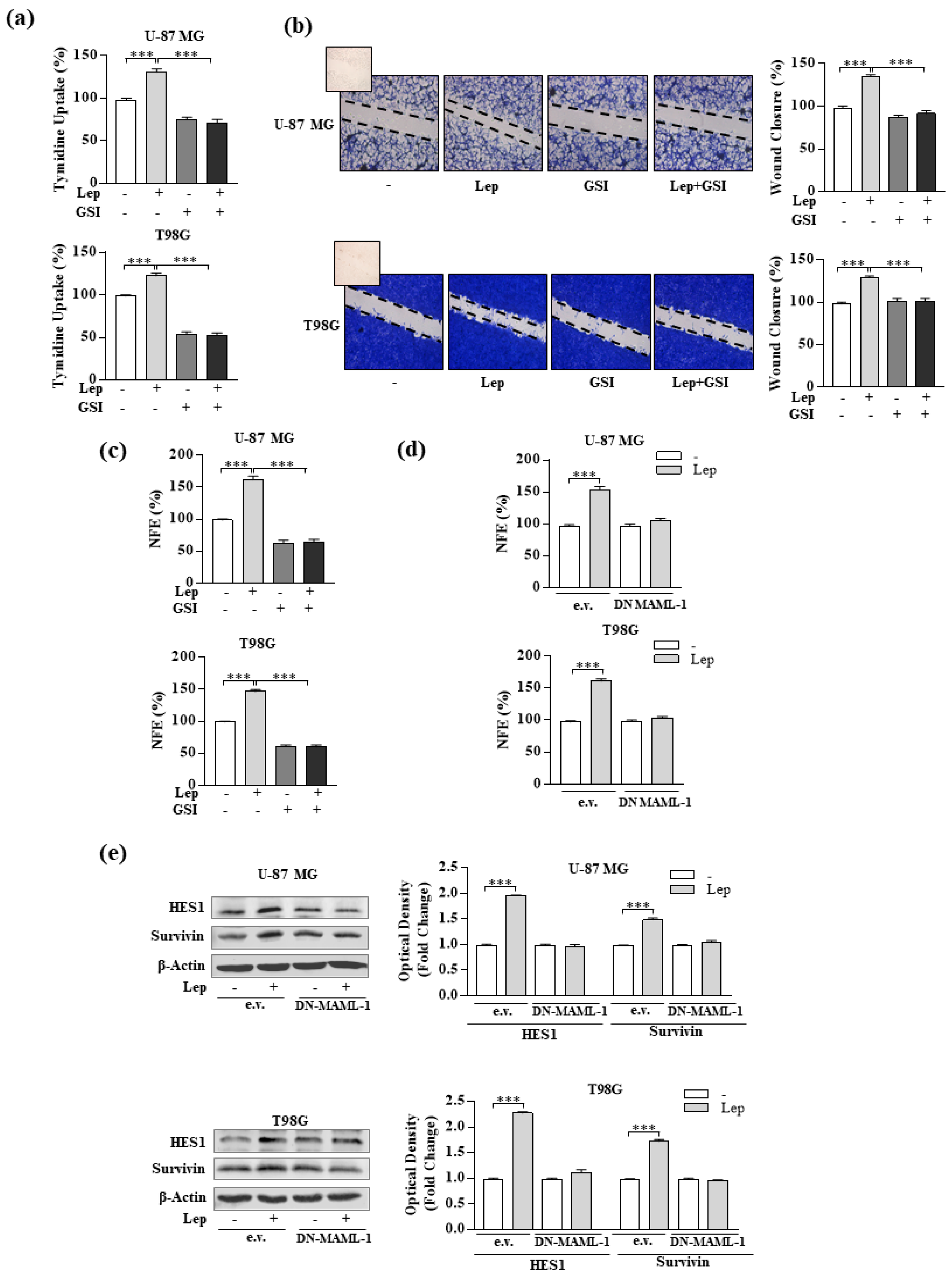
3.4. Interference of Notch Activation Reduces Leptin Effects on Growth and Progression of GBM Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109, Erratum in 2007, 114, 547. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Ann. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef] [Green Version]
- GBD 2016 Brain and Other CNS Cancer Collaborators. Global, regional, and national burden of brain and other CNS cancer, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 376–393. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar]
- Bonavia, R.; Inda, M.M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, 6. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Crean-Tate, K.K.; Reizes, O. Leptin Regulation of Cancer Stem Cells in Breast and Gynecologic Cancer. Endocrinology 2018, 159, 3069–3080. [Google Scholar] [CrossRef]
- Han, G.; Zhao, W.; Wang, L.; Yue, Z.; Zhao, R.; Li, Y.; Zhou, X.; Hu, X.; Liu, J. Leptin enhances the invasive ability of glioma stem-like cells depending on leptin receptor expression. Brain Res. 2014, 1543, 1–8. [Google Scholar] [CrossRef]
- Riolfi, M.; Ferla, R.; Del Valle, L.; Piña-Oviedo, S.; Scolaro, L.; Micciolo, R.; Guidi, M.; Terrasi, M.; Cetto, G.L.; Surmacz, E. Leptin and its receptor are overexpressed in brain tumors and correlate with the degree of malignancy. Brain Pathol. 2010, 20, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.; Wang, L.; Zhao, W.; Yue, Z.; Zhao, R.; Li, Y.; Zhou, X.; Hu, X.; Liu, J. High expression of leptin receptor leads to temozolomide resistance with exhibiting stem/progenitor cell features in gliobalastoma. Cell Cycle. 2013, 12, 3833–3840. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.E.; Cook, N.J.; Rovin, R.A.; Winn, R.J. Leptin promotes glioblastoma. Neurol. Res. Int. 2012, 2012, 870807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, W.L.; Lu, D.Y.; Lee, M.J.; Fu, W.M. Leptin induces migration and invasion of glioma cells through MMP-13 production. Glia 2009, 57, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Wang, L.; Zhao, R.; Yue, Z.; Zhou, X.; Hu, X.; Cao, Y.; Dai, D.; Liu, J. Leptin promotes human glioblastoma growth through activating Signal Transducers and Activators of Transcription 3 signaling. Brain Res. Bull. 2012, 87, 274–279. [Google Scholar] [CrossRef]
- Ferla, R.; Bonomi, M.; Otvos, L., Jr.; Surmacz., E. Glioblastoma-derived leptin induces tube formation and growth of endothelial cells: Comparison with VEGF effects. BMC Cancer 2011, 11, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Vizza, D.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; Amicis, F.; Fuqua, S.A.; Catalano, S.; et al. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol. Oncol. 2013, 7, 379–391. [Google Scholar] [CrossRef]
- Catalano, S.; Mauro, L.; Marsico, S.; Giordano, C.; Rizza, P.; Rago, V.; Montanaro, D.; Maggiolini, M.; Panno, M.L.; Andó, S. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J. Biol. Chem. 2004, 279, 19908–19915. [Google Scholar] [CrossRef] [Green Version]
- Watanobe, H.; Habu, S. Leptin regulates growth hormone-releasing factor, somatostatin, and alpha-melanocyte-stimulating hormone but not neuropeptide Y release in rat hypothalamus in vivo: Relation with growth hormone secretion. J. Neurosci. 2002, 22, 6265–6271. [Google Scholar] [CrossRef] [Green Version]
- Min, D.Y.; Jung, E.; Kim, J.; Lee, Y.H.; Shin, S.Y. Leptin stimulates IGF-1 transcription by activating AP-1 in human breast cancer cells. BMB Rep. 2019, 52, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, G.; Gonzalez-Perez, R.R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Gonzalez-Perez, R.R. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS ONE 2011, 6, e21467. [Google Scholar] [CrossRef] [Green Version]
- Battle, M.; Gillespie, C.; Quarshie, A.; HaLanierrmon, V.; Harmon, T.; Wilson, K.; Torroella-Kouri, M.; Gonzalez-Perez, R.R. Obesity induced a leptin-Notch signaling axis in breast cancer. Int. J. Cancer 2014, 134, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Harbuzariu, A.; Rampoldi, A.; Daley-Brown, D.S.; Candelaria, P.; Harmon, T.L.; Lipsey, C.C.; Beech, D.J.; Quarshie, A.; Ilies, G.O.; Gonzalez-Perez, R.R. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 2017, 8, 7740–7752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley-Brown, D.; Harbuzariu, A.; Kurian, A.A.; Oprea-Ilies, G.; Gonzalez-Perez, R.R. Leptin-induced Notch and IL-1 signaling crosstalk in endometrial adenocarcinoma is associated with invasiveness and chemoresistance. World J. Clin. Oncol. 2019, 10, 222–233. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [Green Version]
- Gürsel, D.B.; Berry, N.; Boockvar, J.A. The contribution of Notch signaling to glioblastoma via activation of cancer stem cell self-renewal: The role of the endothelial network. Neurosurgery 2012, 70, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Hai, L.; Zhang, C.; Li, T.; Zhou, X.; Liu, B.; Li, S.; Zhu, M.; Lin, Y.; Yu, S.; Zhang, K.; et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-κB(p65) pathway. Cell Death Dis. 2018, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Panza, S.; Malivindi, R.; Giordano, C.; Barone, I.; Bossi, G.; Lanzino, M.; Sirianni, R.; Mauro, L.; Sisci, D.; et al. Inhibition of Leydig tumor growth by farnesoid X receptor activation: The in vitro and in vivo basis for a novel therapeutic strategy. Int. J. Cancer 2013, 132, 2237–2247. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Malivindi, R.; Giordano, C.; Gu, G.; Panza, S.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; Panno, M.L.; Andò, S. Farnesoid X receptor, through the binding with steroidogenic factor 1-responsive element, inhibits aromatase expression in tumor Leydig cells. J. Biol. Chem. 2010, 285, 5581–5593. [Google Scholar] [CrossRef] [Green Version]
- Panza, S.; Malivindi, R.; Chemi, F.; Rago, V.; Giordano, C.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Giordano, F.; Andò, S.; et al. Glucocorticoid Receptor as a Potential Target to Decrease Aromatase Expression and Inhibit Leydig Tumor Growth. Am. J. Pathol. 2016, 186, 1328–1339. [Google Scholar] [CrossRef] [Green Version]
- Mauro, L.; Sisci, D.; Bartucci, M.; Salerno, M.; Kim, J.; Tam, T.; Guvakova, M.A.; Ando, S.; Surmacz, E. Shc-alpha5beta1 integrin interactions regulate breast cancer cell adhesion and motility. Exp. Cell Res. 1999, 252, 439–448. [Google Scholar] [CrossRef]
- Panza, S.; Gelsomino, L.; Malivindi, R.; Rago, V.; Barone, I.; Giordano, C.; Giordano, F.; Leggio, A.; Comandè, A.; Liguori, A.; et al. Leptin Receptor as a Potential Target to Inhibit Human Testicular Seminoma Growth. Am. J. Pathol. 2019, 189, 687–698. [Google Scholar] [CrossRef]
- Giordano, C.; Barone, I.; Vircillo, V.; Panza, S.; Malivindi, R.; Gelsomino, L.; Pellegrino, M.; Rago, V.; Mauro, L.; Lanzino, M.; et al. Activated FXR Inhibits Leptin Signaling and Counteracts Tumor-promoting Activities of Cancer-Associated Fibroblasts in Breast Malignancy. Sci. Rep. 2016, 6, 21782. [Google Scholar] [CrossRef]
- Seyfrid, M.; Bobrowski, D.; Bakhshinyan, D.; Tatari, N.; Venugopal, C.; Singh, S.K. In Vitro Self-Renewal Assays for Brain Tumor Stem Cells. Methods Mol. Biol. 2019, 1869, 79–84. [Google Scholar]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, D.; Panno, M.L.; Genchi, G.; Fuqua, S.A.; et al. Combined low doses of PPARgamma and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Leggio, A.; Barone, I.; De Marco, R.; Gelsomino, L.; Campana, A.; Malivindi, R.; Panza, S.; Giordano, C.; Liguori, A.; et al. A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. J. Cell Mol. Med. 2015, 19, 1122–1132. [Google Scholar] [CrossRef]
- Giordano, C.; Gelsomino, L.; Barone, I.; Panza, S.; Augimeri, G.; Bonofiglio, D.; Rovito, D.; Naimo, G.D.; Leggio, A.; Catalano, S.; et al. Leptin Modulates Exosome Biogenesis in Breast Cancer Cells: An Additional Mechanism in Cell-to-Cell Communication. J. Clin. Med. 2019, 8, 1027. [Google Scholar] [CrossRef] [Green Version]
- Morash, B.; Johnstone, J.; Leopold, C.; Li, A.; Murphy, P.; Ur, E.; Wilkinson, M. The regulation of leptin gene expression in the C6 glioblastoma cell line. Mol. Cell Endocrinol. 2000, 165, 97–105. [Google Scholar] [CrossRef]
- Zheng, Q.; Banaszak, L.; Fracci, S.; Basali, D.; Dunlap, S.M.; Hursting, S.D.; Rich, J.N.; Hjlemeland, A.B.; Vasanji, A.; Berger, N.A.; et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr. Relat. Cancer 2013, 20, 797–808. [Google Scholar] [CrossRef]
- Thiagarajan, P.S.; Zheng, Q.; Bhagrath, M.; Mulkearns-Hubert, E.E.; Myers, M.G.; Lathia, J.D.; Reizes, O. STAT3 activation by leptin receptor is essential for TNBC stem cell maintenance. Endocr. Relat. Cancer 2017, 24, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Qiu, M.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H.; Pu, P. The oncogenic roles of Notch1 in astrocytic gliomas in vitro and in vivo. J. Neurooncol. 2010, 97, 41–51. [Google Scholar] [CrossRef]
- Fassl, A.; Tagscherer, K.E.; Richter, J.; Diaz, B.M.; Alcantara Llaguno, S.R.; Campos, B.; Kopitz, J.; Herold-Mende, C.; Herzig, S.; Schmidt, M.H.H.; et al. Notch1 signaling promotes survival of glioblastoma cells via EGFR-mediated induction of anti-apoptotic Mcl-1. Oncogene 2012, 31, 4698–4708. [Google Scholar] [CrossRef]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neurooncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Gelsomino, L.; Panza, S.; Giordano, C.; Barone, I.; Gu, G.; Spina, E.; Catalano, S.; Fuqua, S.; Andò, S. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018, 428, 12–20. [Google Scholar] [CrossRef]
- Pupo, M.; Pisano, A.; Abonante, S.; Maggiolini, M.; Musti, A.M. GPER activates Notch signaling in breast cancer cells and cancer-associated fibroblasts (CAFs). Int. J. Biochem. Cell Biol. 2014, 46, 56–67. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Wood, M.D.; Halfpenny, A.M.; Moore, S.R. Applications of molecular neuro-oncology—A review of diffuse glioma integrated diagnosis and emerging molecular entities. Diagn. Pathol. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-Catalán, M.Á.; Ortuño-Pineda, C.; Moreno-Godínez, M.E.; Villegas-Comonfort, S.; Padilla-Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src-dependent manner in breast cancer cells. Endocr. Connect. 2019, 8, 1539–1552. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, Y.; Funahashi, T.; Tanaka, S.; Taguchi, T.; Tamaki, Y.; Shimomura, I.; Noguchi, S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 2006, 118, 1414–1419. [Google Scholar] [CrossRef]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Zheng, R.; Asano, T.; Nanus, D.M.; Hayakawa, M. Increased serum leptin levels and over expression of leptin receptors are associated with the invasion and progression of renal cell carcinoma. J. Urol. 2006, 176, 1631–1635. [Google Scholar] [CrossRef]
- Candelaria, P.V.; Rampoldi, A.; Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin signaling and cancer chemoresistance: Perspectives. World J. Clin. Oncol. 2017, 8, 106–119. [Google Scholar] [CrossRef]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodol. 2016, 6, 43–55. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Lanier, V.; Gillespie, C.; Leffers, M.; Daley-Brown, D.; Milner, J.; Lipsey, C.; Webb, N.; Anderson, L.M.; Newman, G.; Waltenberger, J.; et al. Leptin-induced transphosphorylation of vascular endothelial growth factor receptor increases Notch and stimulates endothelial cell angiogenic transformation. Int. J. Biochem. Cell Biol. 2016, 79, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. Pt 10J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.P.; Zheng, G.; Zou, L.; Liu, H.L.; Hou, L.H.; Zhou, P.; Yin, D.D.; Zheng, Q.J.; Liang, L.; Zhang, S.Z.; et al. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Mol. Cell Biochem. 2008, 307, 101–108. [Google Scholar] [CrossRef]
- Konishi, H.; Asano, N.; Imatani, A.; Kimura, O.; Kondo, Y.; Jin, X.; Kanno, T.; Hatta, W.; Ara, N.; Asanuma, K.; et al. Notch1 directly induced CD133 expression in human diffuse type gastric cancers. Oncotarget 2016, 7, 56598–56607. [Google Scholar] [CrossRef] [Green Version]
- Bayin, N.S.; Frenster, J.D.; Sen, R.; Si, S.; Modrek, A.S.; Galifianakis, N.; Dolgalev, I.; Ortenzi, V.; Illa-Bochaca, I.; Khahera, A.; et al. Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget 2017, 8, 64932–64953. [Google Scholar] [CrossRef] [Green Version]
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Casalbore, P.; Felsani, A.; Giannetti, S.; Trevisi, G.; Althani, A.; Mangiola, A. The interference of Notch1 target Hes1 affects cell growth, differentiation and invasiveness of glioblastoma stem cells through modulation of multiple oncogenic targets. Oncotarget 2017, 8, 17873–17886. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panza, S.; Russo, U.; Giordano, F.; Leggio, A.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Malivindi, R.; Conforti, F.L.; Naimo, G.D.; et al. Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules 2020, 10, 886. https://doi.org/10.3390/biom10060886
Panza S, Russo U, Giordano F, Leggio A, Barone I, Bonofiglio D, Gelsomino L, Malivindi R, Conforti FL, Naimo GD, et al. Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules. 2020; 10(6):886. https://doi.org/10.3390/biom10060886
Chicago/Turabian StylePanza, Salvatore, Umberto Russo, Francesca Giordano, Antonella Leggio, Ines Barone, Daniela Bonofiglio, Luca Gelsomino, Rocco Malivindi, Francesca Luisa Conforti, Giuseppina Daniela Naimo, and et al. 2020. "Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression" Biomolecules 10, no. 6: 886. https://doi.org/10.3390/biom10060886
APA StylePanza, S., Russo, U., Giordano, F., Leggio, A., Barone, I., Bonofiglio, D., Gelsomino, L., Malivindi, R., Conforti, F. L., Naimo, G. D., Giordano, C., Catalano, S., & Andò, S. (2020). Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules, 10(6), 886. https://doi.org/10.3390/biom10060886