Regulation of Interferon Induction by the Ubiquitin-Like Modifier FAT10



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
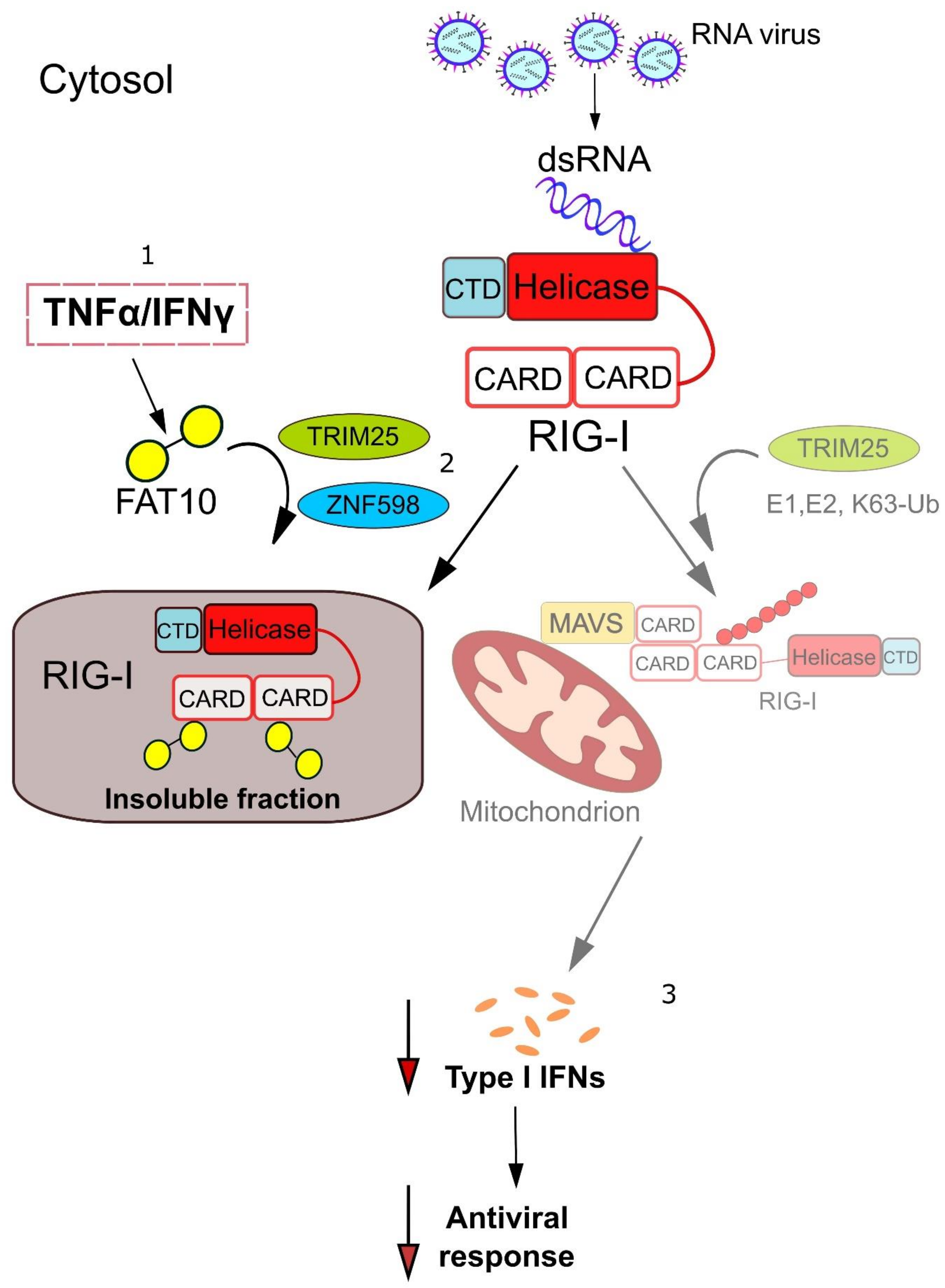{kind=link}
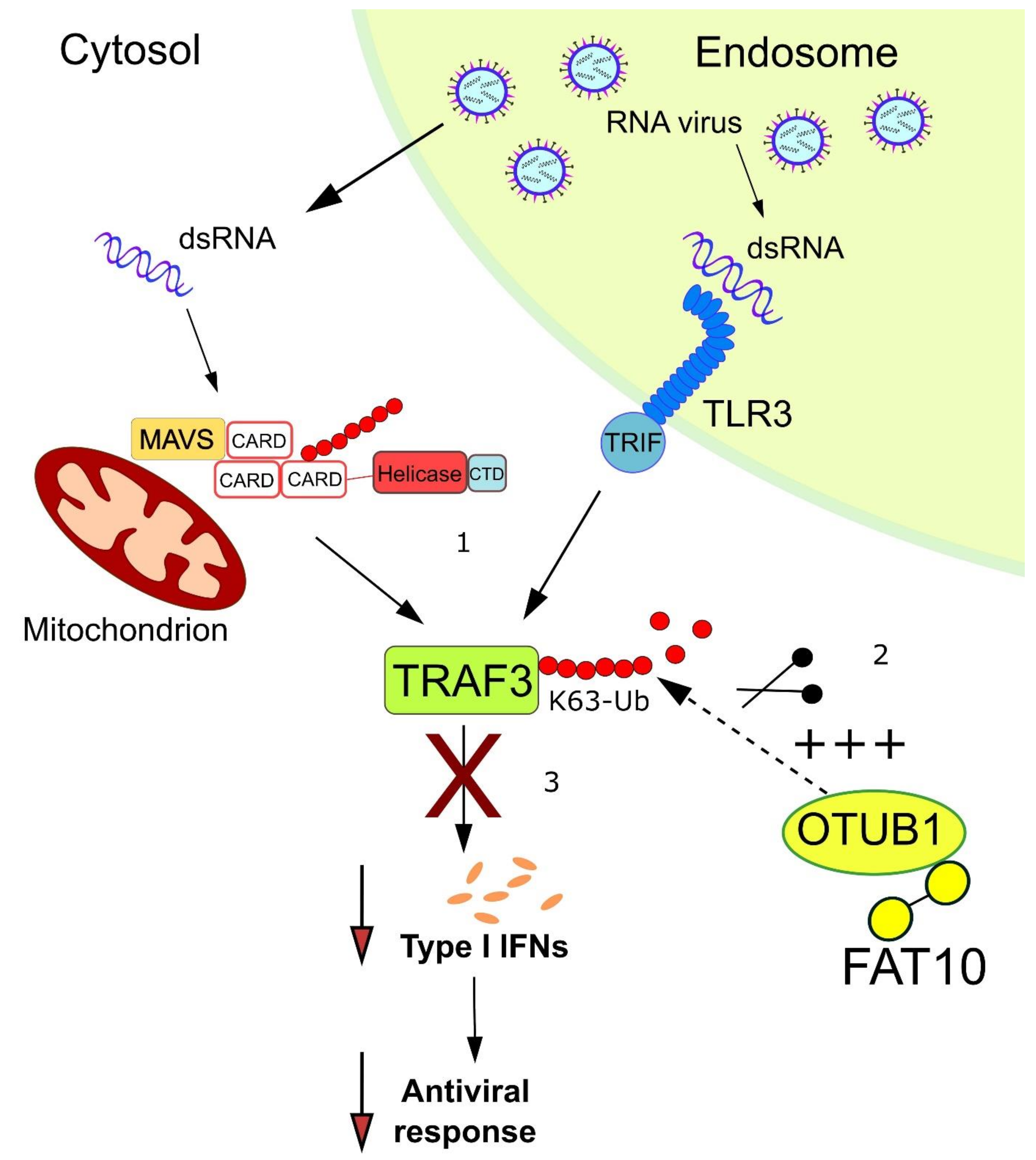{kind=link}
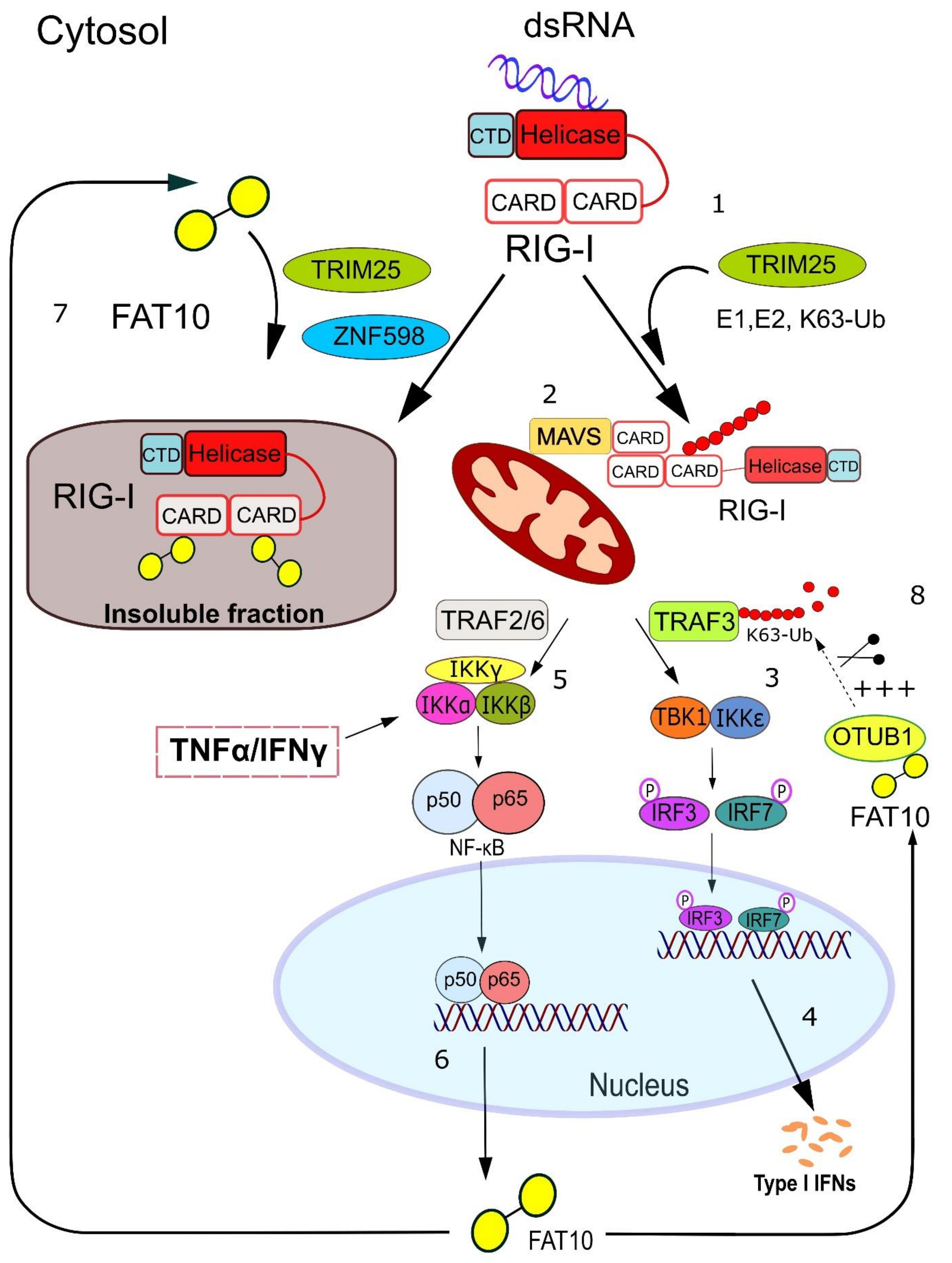{kind=link}
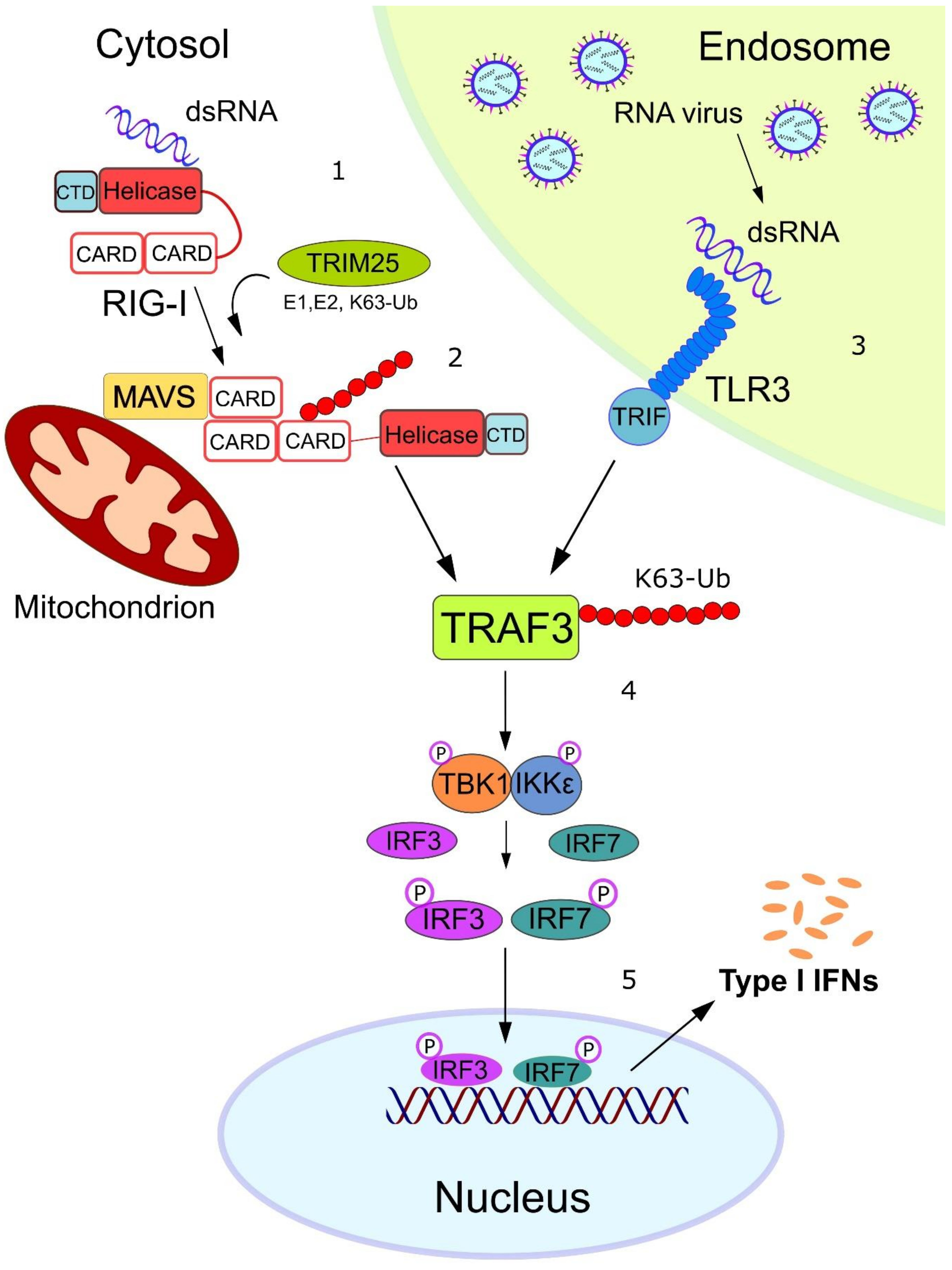
1. Introduction
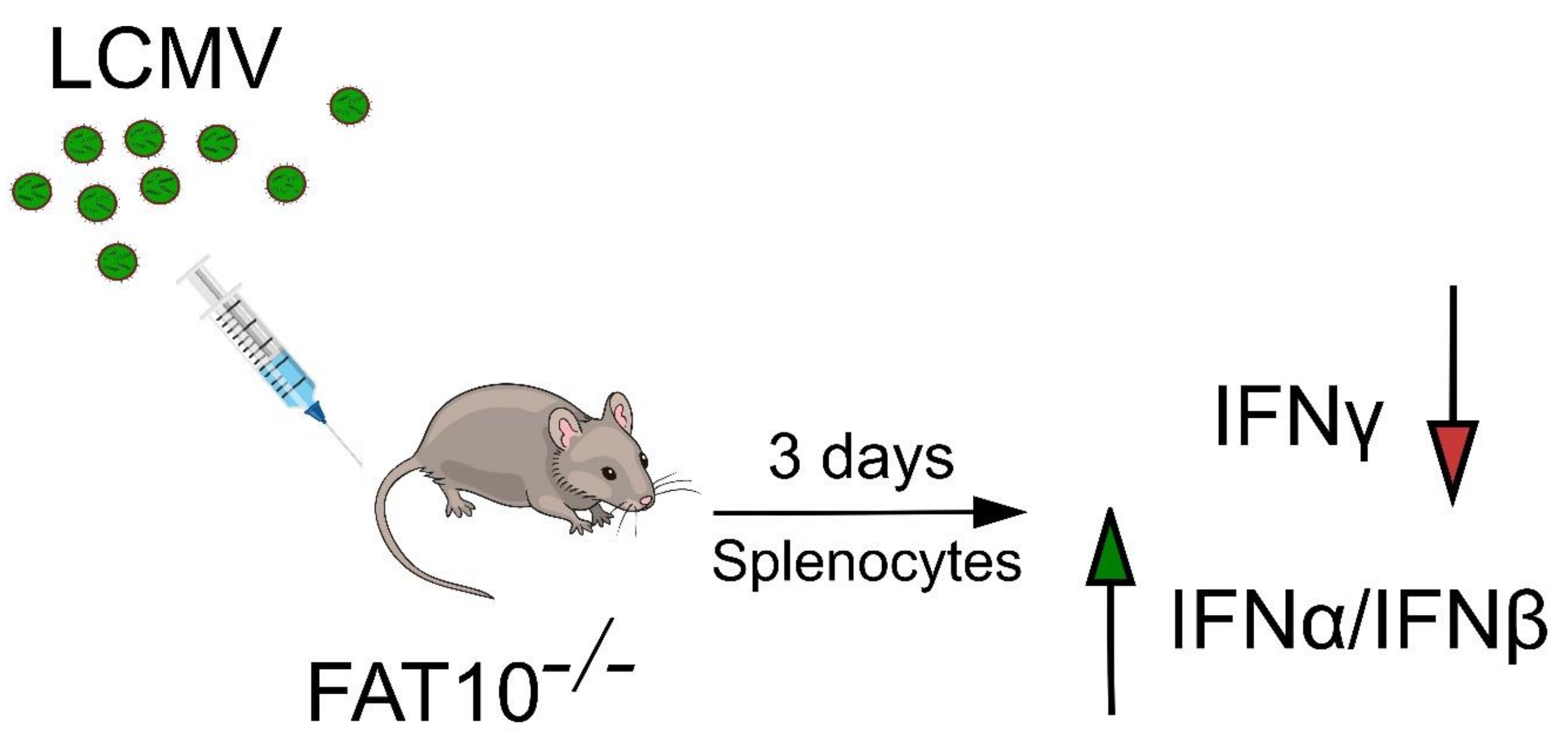
2. FAT10 Modulates IFN Production
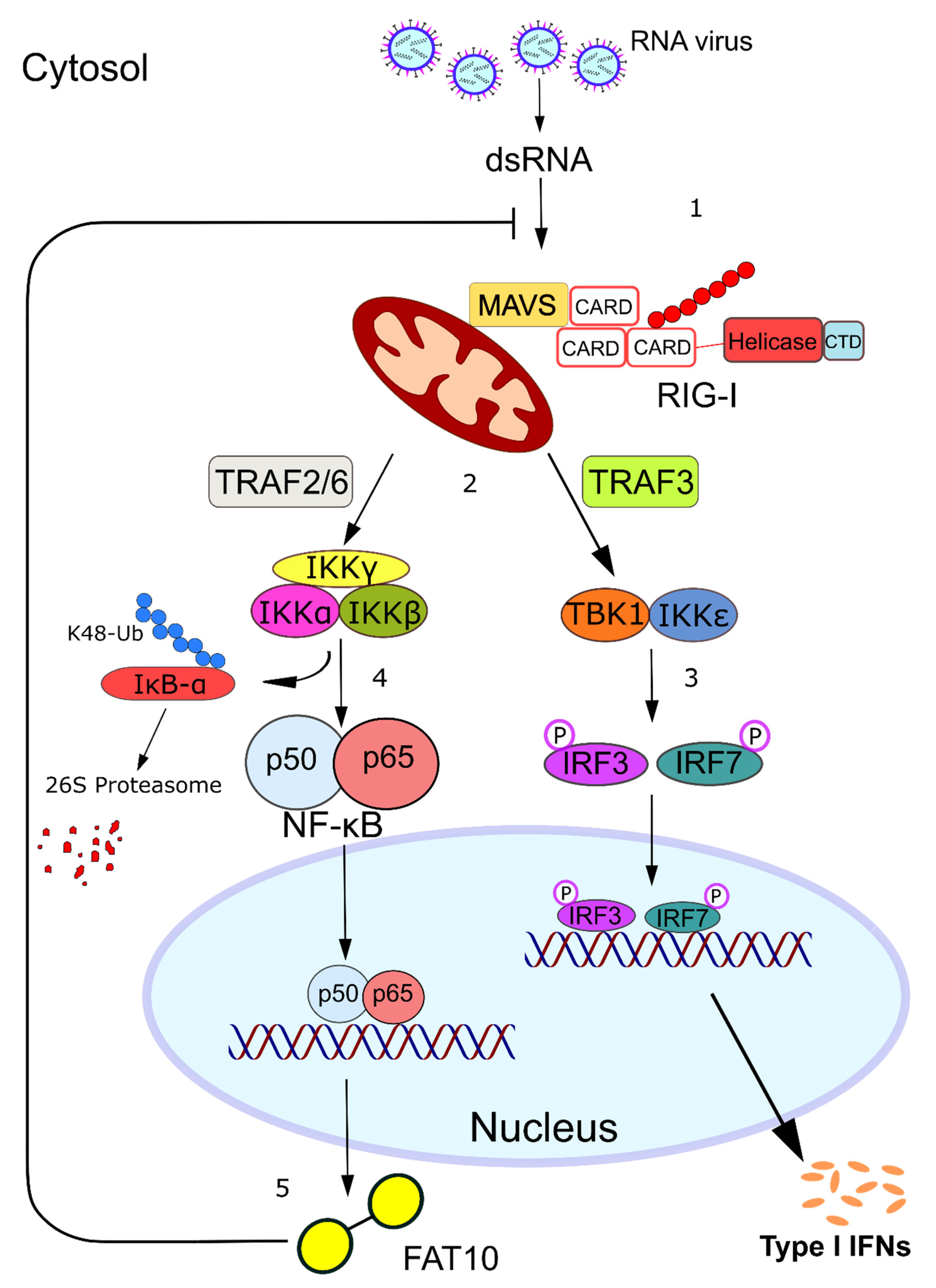
3. FAT10 Impairs the Type I Interferon Signaling Cascade
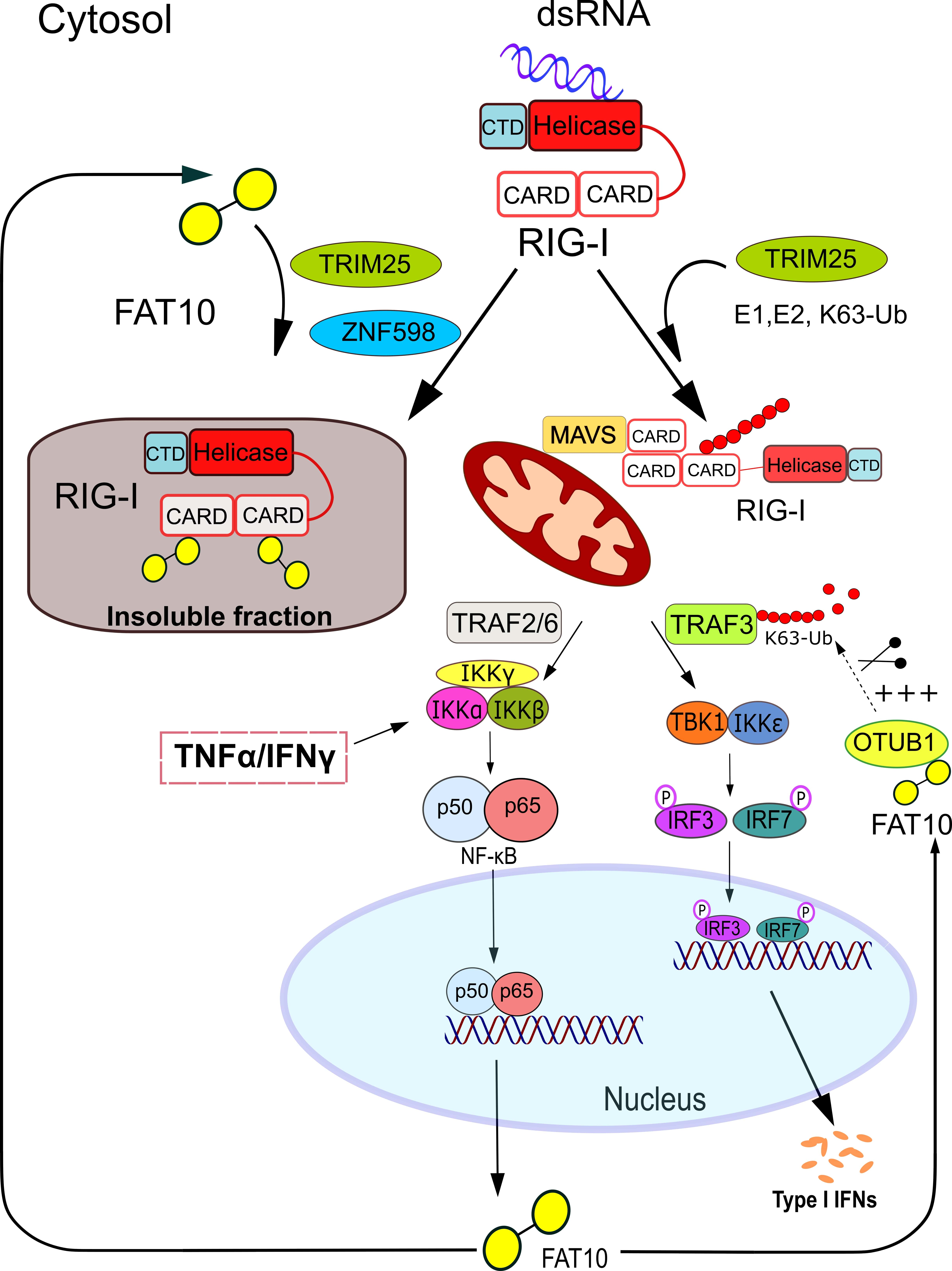
4. Mechanistic Insights into how FAT10 Attenuates RIG-I-Mediated Signal Transduction
5. TRIM25 and ZNF598—Ubiquitin Ligases Involved in the Regulation of RIG-I Signaling by FAT10
6. Further Down the Interferon Induction Cascade: FAT10 Activates the Deubiquitylating Enzyme OTUB1
7. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fan, W.; Cai, W.; Parimoo, S.; Schwarz, D.C.; Lennon, G.G.; Weissman, S.M. Identification of seven new human MHC class I region genes around the HLA-F locus. Immunogenetics 1996, 44, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Pan, J.; Zhang, C.; Fan, W.; Collinge, M.; Bender, J.R.; Weissman, S.M. A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc. Natl. Acad. Sci. USA 1999, 96, 4313–4318. [Google Scholar] [CrossRef] [Green Version]
- Raasi, S.; Schmidtke, G.; de Giuli, R.; Groettrup, M. A ubiquitin-like protein which is synergistically inducible by interferon-gamma and tumor necrosis factor-alpha. Eur. J. Immunol. 1999, 29, 4030–4036. [Google Scholar] [CrossRef]
- Schregle, R.; Mah, M.M.; Mueller, S.; Aichem, A.; Basler, M.; Groettrup, M. The expression profile of the ubiquitin-like modifier FAT10 in immune cells suggests cell type-specific functions. Immunogenetics 2018, 70, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Aichem, A.; Anders, S.; Catone, N.; Rossler, P.; Stotz, S.; Berg, A.; Schwab, R.; Scheuermann, S.; Bialas, J.; Schutz-Stoffregen, M.C.; et al. The structure of the ubiquitin-like modifier FAT10 reveals an alternative targeting mechanism for proteasomal degradation. Nat. Commun. 2018, 9, 3321. [Google Scholar] [CrossRef] [PubMed]
- Theng, S.S.; Wang, W.; Mah, W.C.; Chan, C.; Zhuo, J.; Gao, Y.; Qin, H.; Lim, L.; Chong, S.S.; Song, J.; et al. Disruption of FAT10-MAD2 binding inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2014, 111, 5282–5291. [Google Scholar] [CrossRef] [Green Version]
- Bates, E.E.; Ravel, O.; Dieu, M.C.; Ho, S.; Guret, C.; Bridon, J.M.; Ait-Yahia, S.; Briere, F.; Caux, C.; Banchereau, J.; et al. Identification and analysis of a novel member of the ubiquitin family expressed in dendritic cells and mature B cells. Eur. J. Immunol. 1997, 27, 2471–2477. [Google Scholar] [CrossRef]
- Canaan, A.; Yu, X.; Booth, C.J.; Lian, J.; Lazar, I.; Gamfi, S.L.; Castille, K.; Kohya, N.; Nakayama, Y.; Liu, Y.C.; et al. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol. Cell. Biol. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [Green Version]
- Gruen, J.R.; Nalabolu, S.R.; Chu, T.W.; Bowlus, C.; Fan, W.F.; Goei, V.L.; Wei, H.; Sivakamasundari, R.; Liu, Y.; Xu, H.X.; et al. A transcription map of the major histocompatibility complex (MHC) class I region. Genomics 1996, 36, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Mah, M.M.; Basler, M.; Groettrup, M. The ubiquitin-like modifier FAT10 is required for normal IFN-gamma production by activated CD8(+) T cells. Mol. Immunol. 2019, 108, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kalveram, B.; Raasi, S.; Groettrup, M.; Schmidtke, G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol. Cell Biol. 2005, 25, 3483–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, G.; Kalveram, B.; Groettrup, M. Degradation of FAT10 by the 26S proteasome is independent of ubiquitylation but relies on NUB1L. FEBS Lett. 2009, 583, 591–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, G.; Aichem, A.; Groettrup, M. FAT10ylation as a signal for proteasomal degradation. Biochim. Biophys. Acta 2014, 1843, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, C.; Kassner, I.; Matentzoglu, K.; Singh, R.K.; Wollscheid, H.P.; Scheffner, M.; Schmidtke, G.; Groettrup, M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 2007, 282, 23010–23014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.H.; Sun, Q.; Chen, Z.J. E1-L2 activates both ubiquitin and FAT10. Mol. Cell 2007, 27, 1014–1023. [Google Scholar] [CrossRef]
- Jin, J.; Li, X.; Gygi, S.P.; Harper, J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007, 447, 1135–1138. [Google Scholar] [CrossRef]
- Rani, N.; Aichem, A.; Schmidtke, G.; Kreft, S.G.; Groettrup, M. FAT10 and NUB1L bind to the VWA domain of Rpn10 and Rpn1 to enable proteasome-mediated proteolysis. Nat. Commun. 2012, 3, 749. [Google Scholar] [CrossRef] [Green Version]
- Aichem, A.; Kalveram, B.; Spinnenhirn, V.; Kluge, K.; Catone, N.; Johansen, T.; Groettrup, M. The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J. Cell Sci. 2012, 125, 4576–4585. [Google Scholar] [CrossRef] [Green Version]
- Raasi, S.; Schmidtke, G.; Groettrup, M. The ubiquitin-like protein FAT10 forms covalent conjugates and induces apoptosis. J. Biol. Chem. 2001, 276, 35334–35343. [Google Scholar] [CrossRef] [Green Version]
- Ross, M.J.; Wosnitzer, M.S.; Ross, M.D.; Granelli, B.; Gusella, G.L.; Husain, M.; Kaufman, L.; Vasievich, M.; D’Agati, V.D.; Wilson, P.D.; et al. Role of ubiquitin-like protein FAT10 in epithelial apoptosis in renal disease. J. Am. Soc. Nephrol. 2006, 17, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Merbl, Y.; Refour, P.; Patel, H.; Springer, M.; Kirschner, M.W. Profiling of Ubiquitin-like Modifications Reveals Features of Mitotic Control. Cell 2013, 152, 1160–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.W.; Wang, Y.; Gao, Y.; Mehta, S.B.K.; Lee, C.G.L. FAT10 mediates the effect of TNF-alpha in inducing chromosomal instability. J. Cell Sci. 2011, 124, 3665–3675. [Google Scholar] [CrossRef] [Green Version]
- Gong, P.; Canaan, A.; Wang, B.; Leventhal, J.; Snyder, A.; Nair, V.; Cohen, C.D.; Kretzler, M.; D’Agati, V.; Weissman, S.; et al. The ubiquitin-like protein FAT10 mediates NF-kappaB activation. J. Am. Soc. Nephrol. 2010, 21, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Aichem, A.; Groettrup, M. The ubiquitin-like modifier FAT10 in cancer development. Int. J. Biochem. Cell Biol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canaan, A.; DeFuria, J.; Perelman, E.; Schultz, V.; Seay, M.; Tuck, D.; Flavell, R.A.; Snyder, M.P.; Obin, M.S.; Weissman, S.M. Extended lifespan and reduced adiposity in mice lacking the FAT10 gene. Proc. Natl. Acad. Sci. USA 2014, 111, 5313–5318. [Google Scholar] [CrossRef] [Green Version]
- Buerger, S.; Herrmann, V.L.; Mundt, S.; Trautwein, N.; Groettrup, M.; Basler, M. The Ubiquitin-like Modifier FAT10 Is Selectively Expressed in Medullary Thymic Epithelial Cells and Modifies T Cell Selection. J. Immunol. 2015, 195, 4106–4116. [Google Scholar] [CrossRef] [Green Version]
- Spinnenhirn, V.; Farhan, H.; Basler, M.; Aichem, A.; Canaan, A.; Groettrup, M. The ubiquitin-like modifier FAT10 decorates autophagy-targeted Salmonella and contributes to Salmonella resistance in mice. J. Cell Sci. 2014, 127, 4883–4893. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Q.; Chen, W.; Wang, C. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013, 24, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.G.; Ren, J.; Cheong, I.S.; Ban, K.H.; Ooi, L.L.; Yong Tan, S.; Kan, A.; Nuchprayoon, I.; Jin, R.; Lee, K.H.; et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003, 22, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Lukasiak, S.; Schiller, C.; Oehlschlaeger, P.; Schmidtke, G.; Krause, P.; Legler, D.F.; Autschbach, F.; Schirmacher, P.; Breuhahn, K.; Groettrup, M. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene 2008, 27, 6068–6074. [Google Scholar] [CrossRef] [Green Version]
- Kandel-Kfir, M.; Garcia-Milan, R.; Gueta, I.; Lubitz, I.; Ben-Zvi, I.; Shaish, A.; Shir, L.; Harats, D.; Mahajan, M.; Canaan, A.; et al. IFNgamma potentiates TNFalpha/TNFR1 signaling to induce FAT10 expression in macrophages. Mol. Immunol. 2020, 117, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, J.; Yang, N.; Liu, Q.; Zhang, Q.; Zhang, Y.; Li, N.; Zhao, Y.; Li, S.; Liu, S.; et al. FAT10 Is Critical in Influenza A Virus Replication by Inhibiting Type I IFN. J. Immunol. 2016, 197, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Beagley, K.W.; France, M.P.; Shen, J.; Husband, A.J. Interferon-gamma plays a critical role in intestinal immunity against Salmonella typhimurium infection. Immunology 2000, 99, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Hacker, H.; Tseng, P.H.; Karin, M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 2011, 11, 457–468. [Google Scholar] [CrossRef]
- Garcia-Sastre, A.; Biron, C.A. Type 1 interferons and the virus-host relationship: A lesson in detente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef]
- Aichem, A.; Pelzer, C.; Lukasiak, S.; Kalveram, B.; Sheppard, P.W.; Rani, N.; Schmidtke, G.; Groettrup, M. USE1 is a bispecific conjugating enzyme for ubiquitin and FAT10, which FAT10ylates itself in cis. Nat. Commun. 2010, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Now, H.; Kim, W.J.; Kim, N.; Yoo, J.Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci. Rep. 2016, 6, 23377. [Google Scholar] [CrossRef] [Green Version]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, S.; Bercovich, B.; Ciechanover, A. FAT10 is a proteasomal degradation signal that is itself regulated by ubiquitination. Mol. Biol. Cell 2012, 23, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, S.; Bercovich, B.; Ziv, T.; Ciechanover, A. Modification of the inflammatory mediator LRRFIP2 by the ubiquitin-like protein FAT10 inhibits its activity during cellular response to LPS. Biochem. Biophys. Res. Commun. 2012, 428, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Aichem, A.; Boehm, A.N.; Catone, N.; Schmidtke, G.; Groettrup, M. Analysis of modification and proteolytic targeting by the ubiquitin-like modifier FAT10. Meth. Enzymol. 2019, 618, 229–256. [Google Scholar] [CrossRef]
- Wang, G.; Kouwaki, T.; Okamoto, M.; Oshiumi, H. Attenuation of the Innate Immune Response against Viral Infection Due to ZNF598-Promoted Binding of FAT10 to RIG-I. Cell Rep. 2019, 28, 1961–1970. [Google Scholar] [CrossRef] [Green Version]
- DiGiuseppe, S.; Rollins, M.G.; Bartom, E.T.; Walsh, D. ZNF598 Plays Distinct Roles in Interferon-Stimulated Gene Expression and Poxvirus Protein Synthesis. Cell Rep. 2018, 23, 1249–1258. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nature reviews. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Juang, Y.C.; Landry, M.C.; Sanches, M.; Vittal, V.; Leung, C.C.; Ceccarelli, D.F.; Mateo, A.R.; Pruneda, J.N.; Mao, D.Y.; Szilard, R.K.; et al. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol. Cell 2012, 45, 384–397. [Google Scholar] [CrossRef] [Green Version]
- Bialas, J.; Boehm, A.N.; Catone, N.; Aichem, A.; Groettrup, M. The ubiquitin-like modifier FAT10 stimulates the activity of deubiquitylating enzyme OTUB1. J. Biol. Chem. 2019, 294, 4315–4330. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mah, M.M.; Roverato, N.; Groettrup, M. Regulation of Interferon Induction by the Ubiquitin-Like Modifier FAT10. Biomolecules 2020, 10, 951. https://doi.org/10.3390/biom10060951
Mah MM, Roverato N, Groettrup M. Regulation of Interferon Induction by the Ubiquitin-Like Modifier FAT10. Biomolecules. 2020; 10(6):951. https://doi.org/10.3390/biom10060951
Chicago/Turabian StyleMah, Mei Min, Nicola Roverato, and Marcus Groettrup. 2020. "Regulation of Interferon Induction by the Ubiquitin-Like Modifier FAT10" Biomolecules 10, no. 6: 951. https://doi.org/10.3390/biom10060951
APA StyleMah, M. M., Roverato, N., & Groettrup, M. (2020). Regulation of Interferon Induction by the Ubiquitin-Like Modifier FAT10. Biomolecules, 10(6), 951. https://doi.org/10.3390/biom10060951