1. Introduction
Antibody-based drugs are well accepted for cancer therapy due to the versatility of antibodies for efficacy optimization via antibody engineering. The clinically validated modality of antibodies has been expanding, for example, from whole antibody (IgG) and antibody fragments such as scFv and Fab to antibody–drug conjugates (ADCs) and bispecific antibodies. With recent FDA approval of polatuzumab vedotin (Polivy
®, anti-CD79b antibody conjugated with MMAE payload) for diffuse B cell lymphoma, currently, five ADCs are available in clinics. In the past two years, 230 out of 678 (34%) ADC clinical trials have entered into Phase 2 and Phase2/3, while 62 out of 678 (9%) entered into clinical trials in Phase 3 and Phase 4, further highlighting ADCs as a novel cancer therapeutic strategy [
1].
The cancer cell-killing mechanism of action of an ADC differs from the conventional antibody. The core mechanism of action of an antibody requires the specificity of the antibody against the target antigens and ability to block the interaction between the receptors and its binding partners. This, in turn, intervenes the receptor mediated biological function and downstream signaling pathways that ultimately lead to killing cancer cells as in anti-Her2 targeting antibodies [
2]. The additional efficacy of the antibody for cancer treatment relies on the activation of antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity from FcγR binding to the antibody [
3,
4]. In contrast, ADC consists of a target-specific antibody, a small cytotoxic molecule (payload), and a linker for the conjugation of the payload to the antibody. The potency of a payload primarily contributes to killing cancer cells by ADCs; the antibody of ADC components functions as a drug delivery vehicle for the payload at the targeted site [
5]. Upon ADC binding to the target antigen, the whole molecule of ADC is internalized into cancer cells, followed by the release of the payload from of the antibody. The released payload then exerts its cytotoxic effect on the cancer cells based on the mechanism of action of its cytotoxic payload, such as the cleavage of double-stranded DNA and the inhibition of microtubule formation [
6,
7]. Additional studies have indicated that the intracellular trafficking of ADCs from the endocytic pathway to the lysosomes may provide an improved potency of ADCs that would correlate with an improved clinical response [
8]. Thus, in order to be used as an ADC, the biochemical and biophysical properties of an antibody require the antibody to bind to target cancer cells with specificity and to efficiently and rapidly internalize into a targeted cancer cell while in complex with its interacting partner, then, ultimately, it is delivered to lysosomes for anti-tumor activity.
In recent years, antibody fragment–drug conjugates (FDCs) have been emerging as the next generation of ADCs to improve clinical efficacy, particularly for the unmet need of solid tumors. FDCs are smaller in molecular size than the conventional ADCs and, thus, have a better diffusive penetration ability into tumor tissues. Consequently, an improved rate of cytotoxic drug delivery and increased drug accumulation in the tumor have been observed for FDCs compared with ADCs [
9,
10]. However, the biotherapeutic modalities for FDCs require a molecular size larger than the optimal molecular size for penetration to compensate for their short half-life. FDCs are modified to improve the pharmacokinetics (PK) profile because the standard route of administration is by intravenous injection. On the contrary, the extension of the half-life of FDCs by PEGylation [
11] or fusion with other proteins, such as human serum albumin [
12], leads to an increase in molecular size, which, in turn, reduces drug penetration into tumors. Thus, the efficient internalization of FDC for anti-tumor efficacy is as crucial as it is for ADCs, as demonstrated by the anti-Her2 scFv–HSA–DM1 conjugate [
12]. Therefore, the critical factor in the successful development of ADCs and FDCs is the identification of an antibody that is tumor antigen-specific and endocytic.
The cellular uptake of an antibody is a complicated process associated with many factors, including, but not limited to, target binding, affinity, and the biological mechanism governed by the protein in which the antibody binds [
13]. However, there are conflicts in the published reports on the effect of antibody affinity on the antibody internalization level [
13,
14,
15]. A recent review by Thomas and Balthasar highlighted the determinants and complexity for the high degree of variation in antibody disposition [
13]. Consequently, the molecular mechanism on how the determinants interplay to regulate the antibody internalization is not established. These imply that the “ideal” internalizing antibody for therapeutic development would vary depending on the cancer type, the biological mechanism of the target antigen, and the therapeutic modality of an antibody. Thus, the practical approach for streamlining the selection of the “ideal” internalizing antibody from a pool of antibodies would be based on the “observed” antibody internalization, followed by the subsequent intracellular compartmentalization, such as the lysosome, to function as an ADC and FDC.
The selection of internalizing antibodies against targeted cancer cells for clinical applications often require time-consuming experimental processes. Antibody fragments displayed on phagemid (e.g., phage-displayed scFv antibody libraries) have been mostly utilized in the context of antibody screening for selected therapeutic target-based discovery using recombinant target proteins. Other investigators used the whole cell for biopanning (i.e., cell panning) in an attempt to obtain antibodies against target proteins that are difficult to generate as recombinant proteins or to obtain antibodies recognizing antigens pertaining to native structural features. The panel of phage antibody candidates is then genetically altered to antibody formats (IgG and antibody fragments) followed by the expression and purification of the antibody prior to the analysis of antibody internalization by various methods, including flow cytometry-based assay, radioisotope-labeled antibodies and ADCs, and confocal microscopy [
15,
16,
17,
18,
19,
20]. As an alternative approach to facilitate the identification of internalizing antibodies, the recovery of phage antibodies from the cytosolic fraction of the receptor-expressing cells after receptor-mediated endocytosis has been reported [
21,
22]. However, at present, no systematic approach has been reported as to how the efficiency or the rate of internalization are influenced by and/or correlated with the formats of the antibody.
In this study, we generated the scFv-phage, scFv, and scFv-Fc forms of various antibodies, derived from phage antibodies, to investigate the impact of antibody forms on antibody internalization kinetics. Subsequently, the correlation and the trend of internalization among the antibodies were compared with their antibody forms to determine whether the phage antibody can serve as a predictive screening antibody format for its corresponding antibody fragments. We found that the kinetics of scFv-phage internalization are well-correlated with their scFv and scFv-Fc forms. Our systematic study demonstrates that the analysis of the internalization kinetics of scFv-phage provides an alternative method for assessing antibody fragment internalization. Additionally, the present study demonstrates that endocytic antibodies can be rapidly evaluated from phage antibodies without the production of conventional antibody and antibody fragments. With the latest trends for the application of antibody fragments in the development of FDC and bispecific drug conjugates [
9,
23,
24,
25], a rapid predictive screening and evaluation method for internalizing antibody fragments is, therefore, a useful strategic approach for the selection of optimal antibodies for antibody fragment-based drug development.
2. Materials and Methods
2.1. Cell Culture
HEK293 and MDA-MB-453 (ATCC) were cultured in DMEM (Gibco) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin, unless otherwise stated, at 37 °C, 5% CO2 incubator. CHO-DG44 (ATCC) was maintained in DMEM/F12 (Gibco) supplemented with 10% FBS, 0.1 mM sodium hypoxanthine, 16 μM thymidine, and 100 μg/mL penicillin–streptomycin. Bacterial strains E. coli DH5α and TG1 were used for plasmid DNA preparation and the expression of soluble scFv antibodies, respectively.
2.2. Selection of Target-Binding scFv-Phage Antibodies
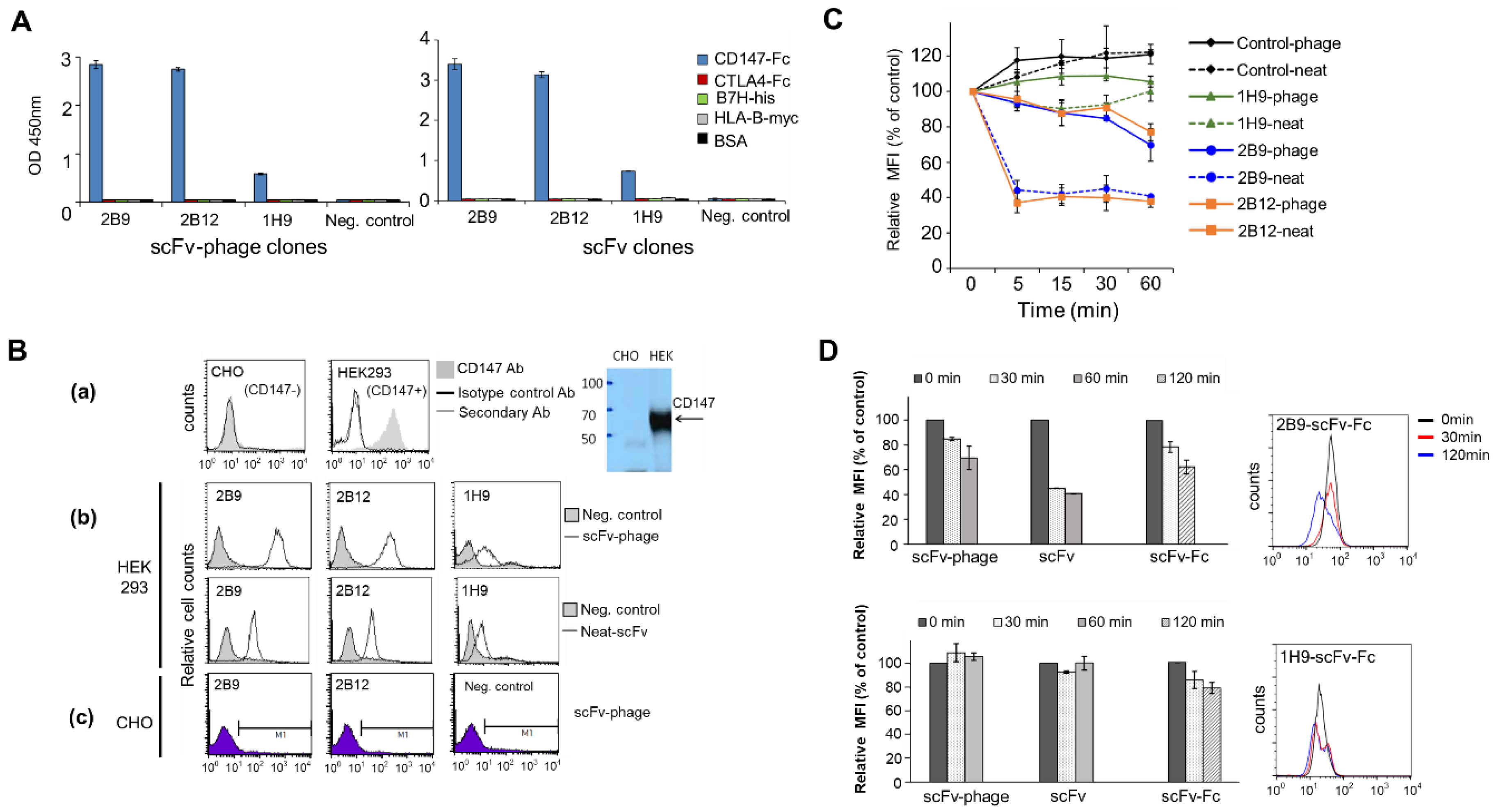
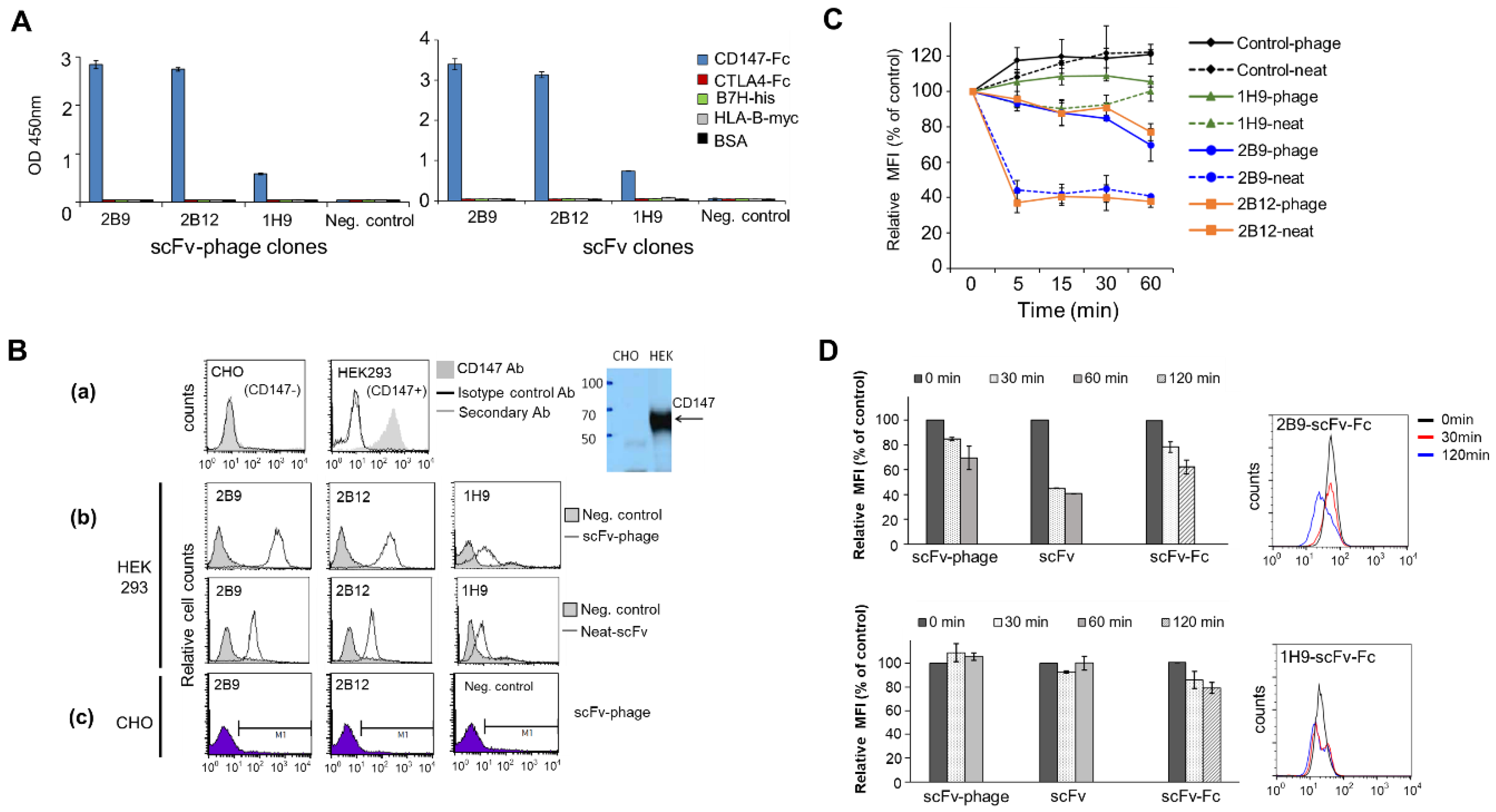
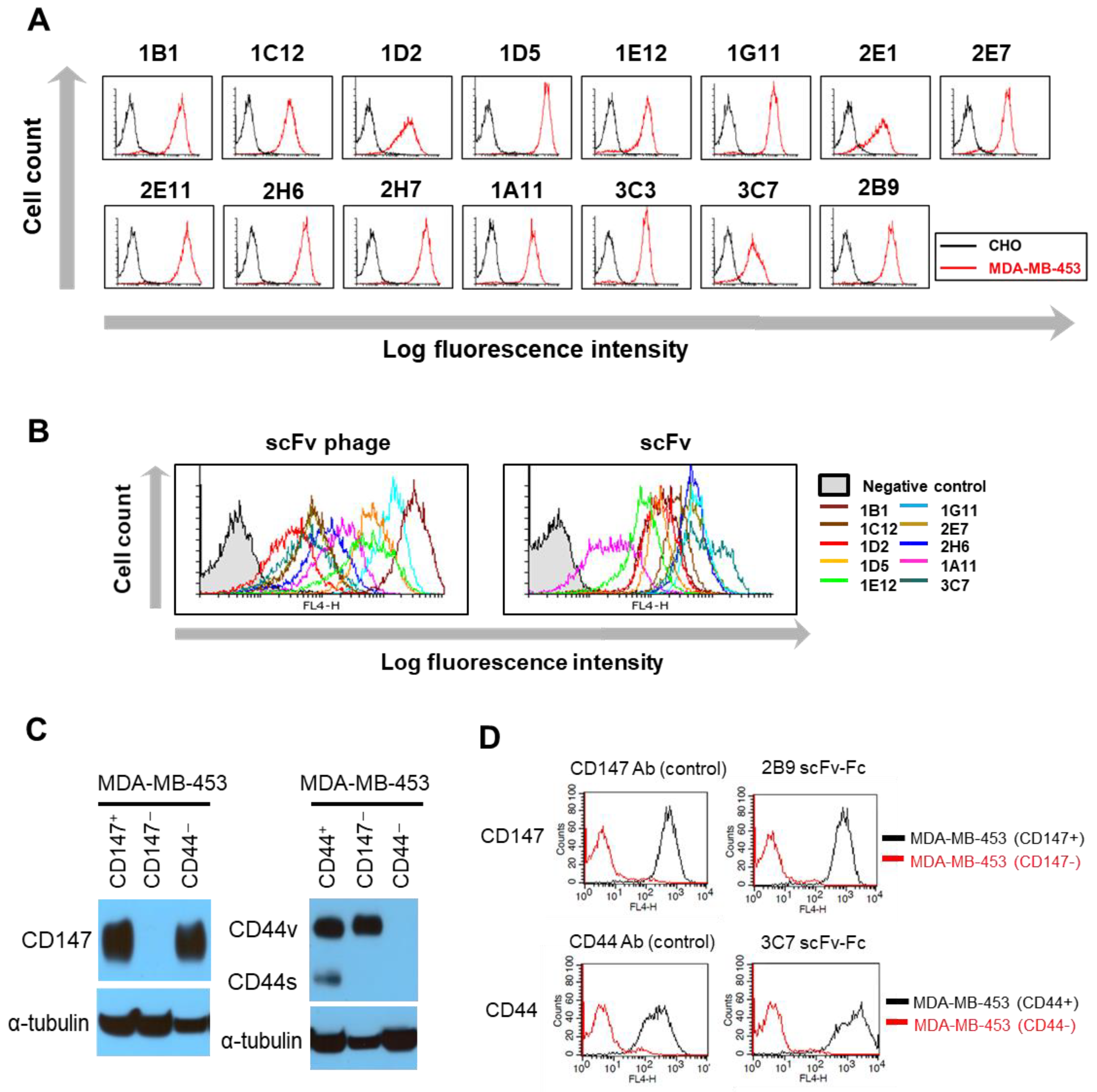
scFv-phage antibodies binding to recombinant human CD147 (Sino Biological Inc.) or to the membrane proteins expressed on the cell surface of MDA-MB-453 (CD44
+/CD133
+/CD24
−) breast cancer (hereafter referred to as MDA-MB-453) cells were selected by biopanning human naïve scFv-phage antibody libraries using standard methods, as previously described, with minor modifications [
26,
27,
28,
29,
30]. The specificity of selected scFv-phage and scFv antibodies for CD147 was analyzed by ELISA using the standard protocol [
29,
30]. BSA and irrelevant proteins with Fc, His or c-Myc tag were used as a negative control. CD147 antibodies bound to the protein coated on the plate were detected using a peroxidase-conjugated anti-M13 phage antibody (Thermo Fisher Scientific) or anti-HA antibody (GenScript) for scFv-phage or scFv, respectively with TMB (Sigma-Aldrich) as the substrate for peroxidase. Absorbance at 450 nm was measured using Victor 4 plate reader (PerkinElmer). The binding of selected scFv-phage antibodies to the cell surface proteins was analyzed by flow cytometry. Briefly, the scFv-phage antibody (1 × 10
9~10 cfu) was incubated with HEK293 for CD147
+ binders or MDA-MB-453 (1 × 10
5 cells) for 1 h at 4 °C. After cell washing with FACS buffer (PBS, 3% BSA, 0.03% NaN
3, pH 7.2), cells were incubated with mouse anti-M13 phage antibody at 1 μg/mL in 100 μL (Thermo Scientific) for 1 h at 4 °C followed by incubation with Alexa Fluor
® 647 conjugated anti-mouse antibody (1:800 dilution, Jackson ImmunoResearch). CHO and PBMC cells were used as a negative controls for CD147
− and for non-cancer cell-surface proteins, respectively. Following washing and re-suspension in PBS containing 4% paraformaldehyde, pH7.4, antibodies bound to the cells were detected and analyzed using a FACSCalibur flow cytometer and CellQuest Pro software (BD Bioscience) (
Figure S1).
2.3. Production of scFv-Phage, Neat-scFv, scFv and scFv-Fc Antibodies
scFv-phage antibodies were prepared as per standard protocol [
29,
30]. Briefly,
E. coli TG1 cells harboring the scFv-phage antibody were grown to the mid-log phase in 2xTY containing 2% glucose and antibiotics (20 µg/mL chloramphenicol or 100 µg/mL ampicillin) with rapid shaking at 37 °C. Helper phage (VCS-M13, Stratagene) was then added and incubated for an additional 1 h with gentle shakings, followed by an exchange of the growth medium containing 50 µg/mL kanamycin in place of glucose. Cells were grown overnight with rapid shaking at 25 °C to produce the scFv-phage antibody. The concentration of the phage antibody in the supernatant by precipitation was performed by the addition of 3/10 volume of precipitation buffer (20%
w/v PEG8000 and 2.5M NaCl) and incubation on ice for at least 2 h, followed by centrifugation at 44,000×
g for 30 min at 4 °C. The pelleted scFv-phage was re-suspended in 10 mM Tris-HCl, pH7.5. The titer of scFv-phage was determined by the infection of the phage into
E. coli TG1 following the standard protocol [
31].
To obtain neat scFv (unpurified crude scFv) by periplasmic extraction, E. coli TG1 cells harboring the scFv with c-Myc/His (MDA-MB-453-binding antibodies (Abs)) or HA/His tag (CD147 Abs) at the C-terminus in the phagemid vector were cultured as described above for scFv-phage, with the following modifications. Cells were grown to the log-phase in medium containing 0.1% glucose at 30 °C, and induced antibody expression with 1 mM IPTG. Cells were pelleted at 3700× g for 10 min after overnight growth with rapid shaking at 25 °C and were then resuspended in ice-cold TES buffer (50 mM Tris-HCl, 1 mM EDTA, 0.5 M sucrose, pH8). Subsequently, a 1.5× volume of 1:5 dilution of TES buffer in cold H2O was added and incubated on ice for 30 min. Cell debris was pelleted at 19,000× g, 4 °C, and the periplasmic extract containing neat scFv was used for subsequent analyses such as ELISA and flow cytometry.
scFv antibodies were prepared by purification of the neat-scFv using anti-c-Myc immunoaffinity (Pierce) for c-Myc-tagged scFv or an immobilized metal ion affinity column (GE Healthcare) for His-tagged scFv, according to the manufacturer’s instructions, on ÄKTA FPLC (GE Healthcare). c-Myc-tagged scFv bound to the column was eluted with 75 mM citric acid, 50 mM NaCl, pH3.0 and immediately neutralized using 1 M sodium phosphate, pH 8. His-tagged scFv bound to the column was washed with 50 mM sodium phosphate, 150 mM NaCl, 20 mM imidazole, pH 7.2. After several washing steps with imidazole, scFv was eluted with 50 mM sodium phosphate, 150 mM NaCl and 200 mM imidazole, pH 7.2.
scFv-Fc antibodies were generated by reformatting the scFv fused to the Fc fragment of human IgG1, and sub-cloned into the mammalian expression vector pCEP4 (Invitrogen). scFv-Fc antibodies were transiently expressed in HEK 293F (Invitrogen) following the transfection of corresponding pCEP4/scFv-Fc expression vectors with polyethyleneimine (PEI). The conditioned medium containing scFv-Fc were collected after cells were cultured for 6–7 days in Freestyle 293 Expression Medium (Invitrogen), followed by purification using HiTrap Protein A or HiTrap MabSelect SuRe (GE Healthcare) following the manufacturer’s instructions. Purified antibody fragments were analyzed by SDS-PAGE (
Figure S2). Antibody concentrations (scFv and scFv-Fc) were determined from the measurement of absorbance at 280 nm, using the calculated extinction coefficient based on the amino acid sequence of the antibody.
2.4. Flow-Based Antibody Internalization Assay
Prior to the evaluation of the antibody for internalization, each antibody binding to the cells was confirmed by flow cytometry. scFv-phage binding to cells was confirmed as described above under the section of “Selection of Target-Binding scFv-Phage Antibodies”. scFv (neat and purified scFv) and scFv-Fc binding to the cells were performed as described in the aforementioned section, except for the mouse anti-c-Myc antibody (5 μg/mL) with Alexa Fluor® 647-labeled anti-mouse Fc-specific F(ab’)2 antibody for scFv and Alexa Fluor® 647-labeled anti-human Fc-specific F(ab’)2 antibody (1:800 dilution, Jackson Immunoresearch) for scFv-Fc, which were used for the detection of the antibody bound to the cell surface.
The internalization of antibodies into cells was analyzed by flow cytometry as described above, except for the following modifications. HEK293 or MDA-MB-453 (1 × 10
5 cells) grown to 80–90% confluence were incubated with scFv-phage (total 10
9 cfu), neat scFv (final 1:3 dilution), scFv (final at 1.25 μg/mL) or scFv-Fc (1.25 μg/mL) as described above. Following cell wash with PBS, 3% BSA, pH 7.4, to remove unbound antibodies, cells were re-suspended with PBS, 1% BSA, pH 7.4, in 400 μL and incubated at 37 °C to allow the antibody to internalize, while the corresponding control antibody samples were incubated at 4 °C to prevent the internalization of the antibody. The samples were collected at various time points (15, 45, and 90 min) by adding 1 mL of pre-chilled cold FACS buffer and incubating them on ice to prevent further internalization and then processing cells for flow cytometry as described above. The percentage of internalization of a cell surface-bound antibody within the cells was determined by comparing the net decrease in mean fluorescence intensity (MFI) of samples incubated at 37 °C to the corresponding control samples incubated at 4 °C. The relative MFI of the sample at each time point x (t = x) to the percentage of the control was calculated as follows:
where MFI
t = 0 is the MFI of the control at 4 °C and MFI
t = x is the MFI of the matching sample at 37 °C at the indicated time point x.
The percentage of Ab internalization at each time point x was then calculated as
2.5. Statistical Analysis
A two-tailed Pearson correlation analysis was performed using SPSS (IBM, New York, NY, USA) for the correlation of internalization among various antibody formats. A Gaussian distribution assessment was carried out via a Shapiro–Wilk test using SPSS, and a t-test was applied using Prism 8 (GraphPad software). The significance of the observed differences in the relative % internalization between the two time points for each antibody clone was tested by using a one-tailed Welch’s t-test. The significance of differences in the relative % internalization for the variation of antibody concentrations or the valency of the antibody were analyzed using unpaired and two-tailed Student’s t-tests. Differences were considered as statistically significant and were annotated as follows: p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001.
2.6. Immunofluorescence-Based Antibody Internalization Assay
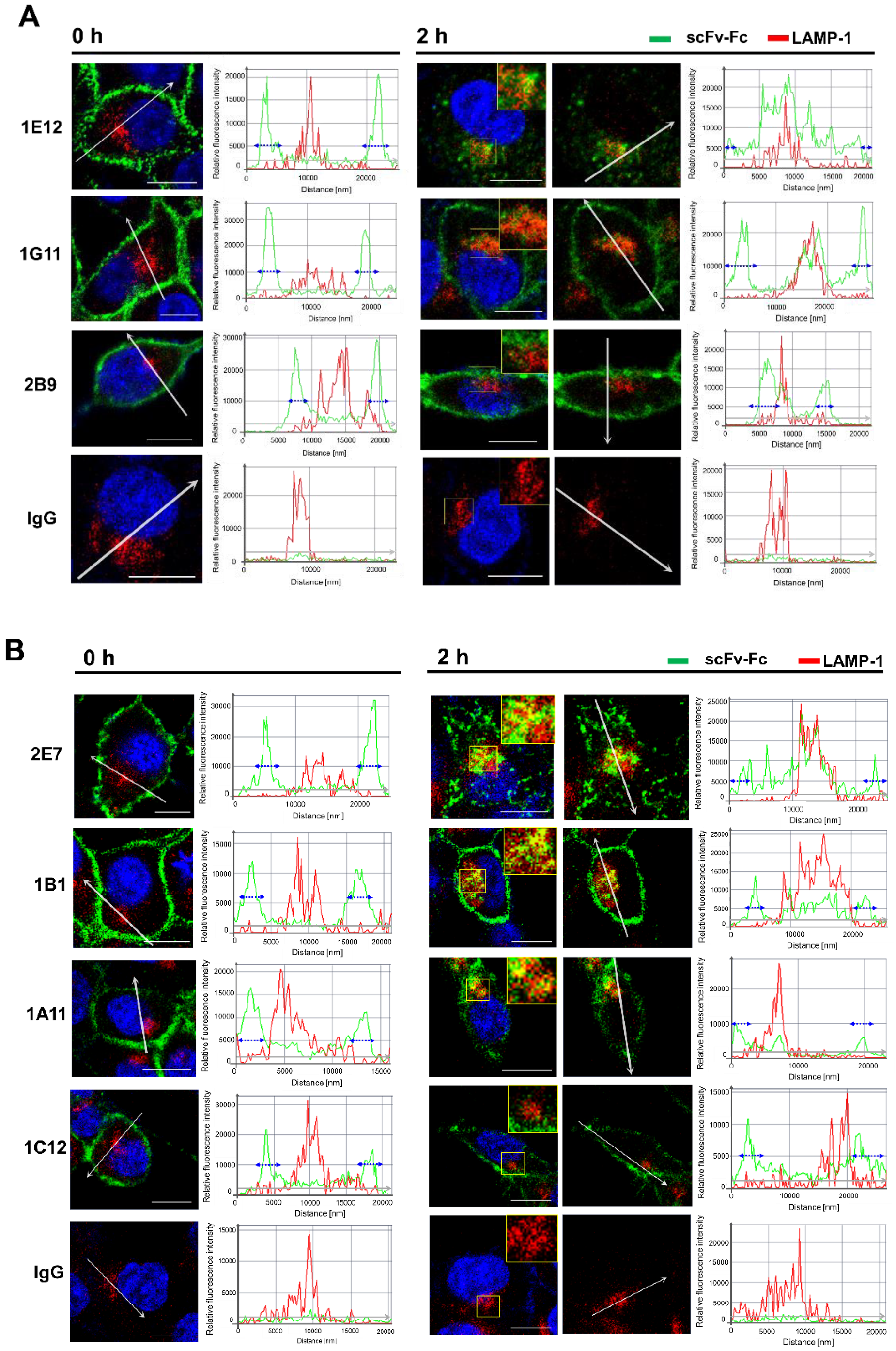
MDA-MB-453 cells grown to 70–80% confluence on a confocal imaging dish (SPL) were washed twice with 1 mL pre-chilled PBS and incubated with scFv-Fc (10 µg/mL) or scFv-phage (total 2.5 × 1010 cfu) on wet ice for 1 h. After the removal of unbound antibodies by an extensive wash with PBS, cells were supplemented with 1 mL of serum-free growth medium followed by incubation for 0 or 2 h at 37 °C. Cells were then washed once with cold PBS and fixed with 1 mL of 4% (w/v) paraformaldehyde for 20 min at RT, followed by two washes with PBS. Cells were then permeabilized with 1 mL of 0.1% of Triton X-100 for 5 min and washed twice with 1 mL of PBS. The subcellular localization of scFv-Fc was detected with 2.5 µg/mL Alexa Fluor® 594-labeled F(ab’)2 anti-human IgG, and scFv-phage was detected with mouse anti-M13 phage antibody followed by 3 µg/mL Alexa Fluor® 594-labeled F(ab’)2 anti-mouse IgG. Lysosomes were stained with 5 µg/mL mouse anti-LAMP-1 antibody (BioLegend, San Diego, CA, USA) or rabbit anti-LAMP-1 antibody (Abcam, Cambridge, USA) for the co-localization analysis of scFv-Fc or scFv-phage, respectively. Alexa Fluor® 647-labeled F(ab’)2 anti-mouse IgG (1:500 dilution, Jackson ImmunoResearch, West Grove, FL, USA) or Alexa Fluor® 647-labeled anti-rabbit IgG (1:500 dilution, Jackson ImmunoResearch) was used for the detection of the anti-LAMP-1 antibody. Images of stained cells were taken with a Zeiss LSM 880 confocal microscope (Carl Zeiss, Pleasanton, CA, USA) with a 40× water immersion objective and processed using ZEN 3.1 software (Carl Zeiss).
2.7. Confocal Analysis for Quantification of Internalized Antibodies
The relative number of internalized antibodies was quantified based on the change in the MFI from flow cytometry and the integrated fluorescence density from the images of stained cells using the method adapted by Gottstein et al. and Marre et al. [
32,
33]. Randomly selected images of cells were analyzed using Zen 3.1 (Carl Zeiss). The cell membrane was identified using all fluorescence channels in Zen 3.1 to distinguish the inside and outside areas of the cell. The ratio of antibody taken up by the cells, which defines the internalization in percent, was calculated as follows:
where F
in and F
out are the integrated fluorescence density of the antibody from inside and outside the cell, respectively, from the cell images.
The integrated fluorescence density, F, is the product of the selected area and the MFI of the selected area from the cell images, and is derived as follows:
where ∑A
in is the sum of the inside area of the cells and ∑MFI
in is the sum of the MFI of the inside area of the cells, and MFI
neg is the mean fluorescence intensity of the negative control (from the secondary antibody only).
where ∑A
out is the sum of the outside area of the cells, ∑MFI
out is the sum of the MFI of the outside area of the cells, and MFI
background is the MFI from the cell-absent area.
Then, the relative amount of antibody taken up by per cell was quantified by:
where MFI
4 °C and MFI
neg.con are the MFI of Abs incubated at 4 °C (before internalization) and the MFI of the negative control (without the primary antibody), respectively, from the flow cytometry analysis.
The number of antibodies bound on the cell surface of each cell can be obtained by [(MFI
4 °C − MFI
neg.con)/MFI of one antibody molecule]. Since the same fluorescent-dye conjugated antibody is used for each set of experiments, the relative number of antibodies bound on each cell surface was obtained using the arbitrarily assigned value for the MFI of one molecule of antibody. The relative number of internalized antibodies in each cell is calculated from the internalization (%) multiplied by the number of antibodies bound to each cell before endocytosis. Uncertainties and error analyses of calculation for internalization (%) were represented as the percentage of standard deviations calculated using the arithmetic calculation of the propagation of error, as noted by Gottstein et al. [
32].
4. Discussion
ADCs, a tumor-specific Ab in complex with a potent cytotoxic payload, have emerged as an effective anti-cancer therapeutic biomolecule. As the number of ADCs entering the clinic increases, the number of innovative antibody structures for drug conjugates, such as FDCs and bispecific Ab–drug conjugates, are also increasing with an additional clinical benefit. The anti-tumor efficacy of the antibody-based drug conjugates is influenced by the internalization efficiency of antibodies towards targeted cancer cells and directing to the lysosomes [
36,
37]. FDCs were developed as the next generation of ADCs to overcome antibody uptake in vivo [
9,
23,
24,
38]. Paradoxically, the recent development of FDCs has focused on improving PK via the re-attachment of a larger molecule (e.g., PEG or HSA), which diminishes the advantage of rapid tumor accumulation and the enhanced penetration of FDCs [
11,
39,
40]. Thus, the identification and evaluation of antibodies possessing optimal rates and high levels of internalization are essential in the development of therapeutic ADCs and FDCs. Various approaches have been employed in an attempt to improve the selection of internalizing antibodies. Biopanning on tumor cells for the selection of phenotypic antibodies and internalizing antibodies have been reported [
21,
41]. However, with the complexity of the phage antibody pool from the cell panning due to the diversity of the cell surface proteins exemplified by Ljungars et al. [
41], the subsequent screening of antibodies is often hampered due to difficulties in obtaining the membrane protein in sufficient quantity and quality, as well as the need to generate antibodies from the phage antibodies.
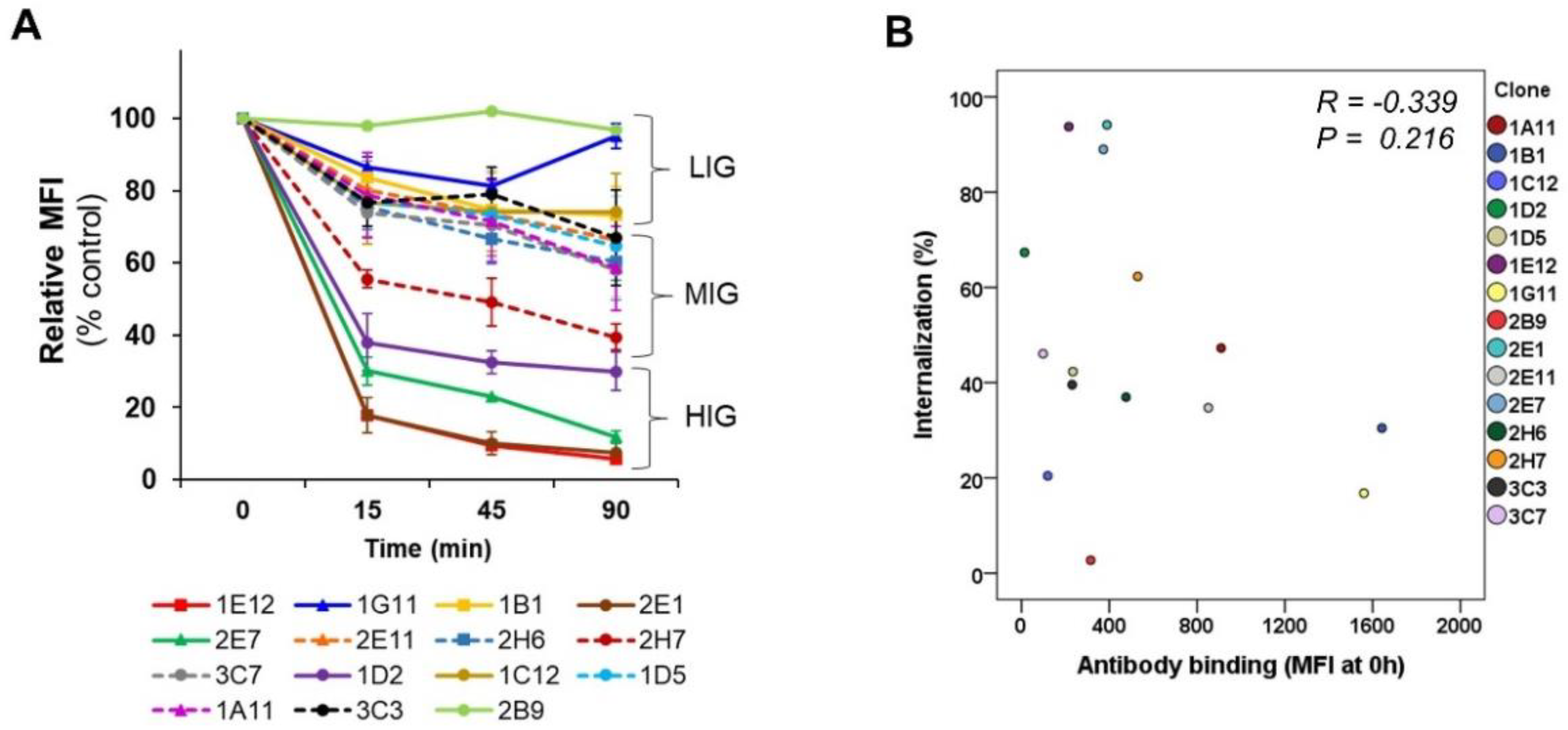
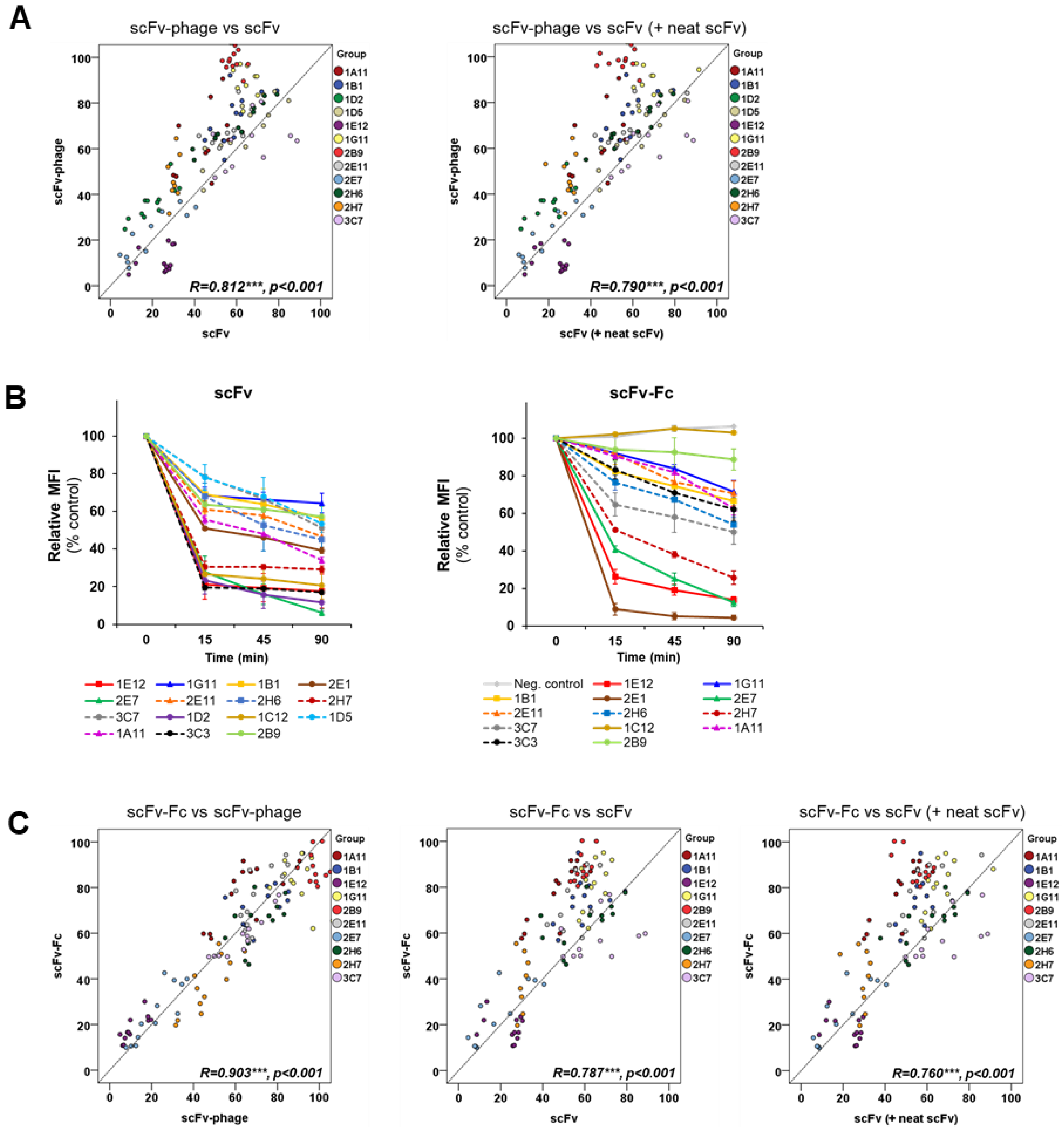
In this study, we sought to determine the relationship between the internalization trend and antibody formats and to investigate whether the optimal internalizing antibody can be screened from phage antibodies without generating full IgG and antibody fragments. Specifically, we analyzed the kinetics of the internalization of a panel of antibodies with a broad range of antigen binding patterns against MDA-MB-453 cells. We performed systematic and side-by-side comparative studies using scFv-phage, scFv, and scFv-Fc fragments to decipher the trend of relative ranking of internalization among the panel of antibodies. Overall trends for the relative kinetics and degrees of scFv-phage internalization were directly correlated with their corresponding scFv and scFv-Fc antibody fragments. As anticipated, an increased amount of antibody taken up by the cells as a function of time was observed. Importantly, the relative order and the endocytosis pattern of phage antibody remained the same for all antibodies: all Abs arbitrarily assigned to the HIG, MIG, or LIG group remained in the same group even after Abs were allowed to internalize for 90 min compared to the internalization level at 15 min.
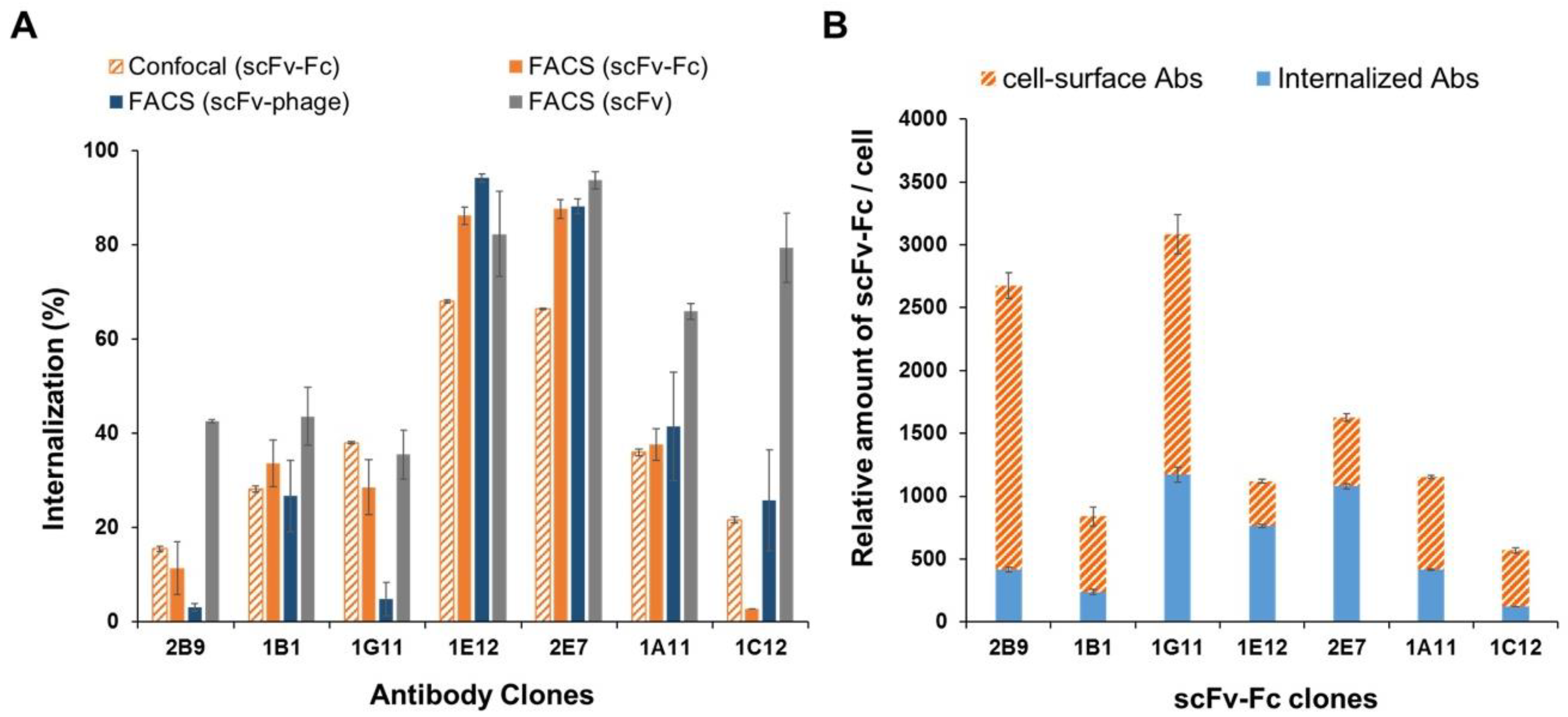
The antibodies in the panel had distinct interactions with their antigens. The initial phage antibody binding to the protein was different by up to tenfold relative to the MFI value. Nonetheless, the comparative evaluation across the scFv-phage, scFv, and scFv-Fc antibody forms consistently demonstrated that the overall trend of total cellular uptake of antibodies does not change significantly. Likewise, the high level of initial Ab binding to the membrane protein did not correlate with the Ab internalization level (
Figure 3B and
Figure S4). For example, 1E12, 1G11, and 2B9 clones have relatively high initial binding to the cells, but 1E12 internalize well, whereas both 1G11 and 2B9 are relatively internalized lower amounts than 1E12 (
Figure 6A). Furthermore, statistical analysis showed that the initial antibody–protein interaction did not influence antibody internalization in all antibody forms (
Figure 3B and
Figure S4). Thus, we assumed that the changes in the internalization level of an antibody among scFv-phage, scFv, and scFv-Fc are likely due to alterations in the antibody format.
The antibodies’ binding affinity and receptor crosslinking can have effect on antibody internalization. Interestingly, scFv-phage and scFv-Fc displayed a better correlation of internalization properties compared with the correlation between scFv-phage and scFv forms. Since we have not used hyperphage for high-valence display, the scFv-phage antibodies used in this study are derived from low-valence phage antibodies (i.e., monovalent). This observation suggests that the valency of antibodies did not alter the relative internalization trend, although the total amount of antibody uptake by cells may differ between monovalent and bivalent antibodies. The importance of the crosslinking of the target receptor, such as FGFR1 and Her2, by the antibody for an efficient internalization has been reported [
42,
43]. In contrast, the efficient internalization of Fab and IgG antibodies, such as anti-EphA2 and anti-CD73, has been observed [
14,
15]. We also observed that the scFv and scFv-Fc antibodies, at an equivalent stoichiometry to the antigens, had a minimum effect on the relative antibody internalization profile. These observations indicate that the influence of receptor clustering on internalization is negligible for the antibodies investigated in this work. The scFvs showed a faster internalization kinetic profile than their scFv-Fc and scFv-phage antibodies. The smaller molecular size of scFv may have contributed to the faster internalization kinetics compared with the scFv-Fc and scFv-phage forms. However, scFv fragments also displayed a range of kinetics of internalization among the scFv antibodies. The trend of the overall internalization profile was consistent with their scFv-Fc and scFv-phage forms. This further demonstrates the consistent trend of rates and levels of internalization across the three antibody forms investigated in this study.
In contrast, we have not observed an indication for antibody internalization associated with the binding patterns (MFI) of an antibody on the cells. We have not yet evaluated the effect of the affinity of each antibody for the antigen on the internalization. Investigations on the effect of antibody affinity in terms of internalization demonstrated the difficulties in correlating the optimal affinity, which would enable predictions for the selection of an “ideal” antibody for internalization. For example, the higher the affinity of the antibody for the antigen is, the higher the efficiency in internalization is for Her2 and FGFR1 antibodies that bind the same epitope on the targeted protein [
44,
45]. In contrast, such significant correlations of affinity or valency of the antibodies in relation to internalization were not observed with CEA antibodies that had either the same or different epitopes from each other in monovalent or bivalent antibody form [
46]. Furthermore, the affinity of the antibody is thought to be more important for low-density receptors to be taken up by the tumor cell [
45,
47]. Thus, it would be interesting to follow up on how the affinity of these antibodies influences internalization in the subsequent studies.
The trend of internalization was consistent by both flow cytometry-based and confocal image analysis for most of the antibodies in the panel, regardless of the format of these antibodies. Moreover, the subcellular co-localization of antibodies demonstrated that the Abs are taken up by the endocytic processes and shuttled into the intracellular lysosome compartment. Moreover, the trends of the rates and levels of internalization for scFv and scFv-Fc antibody fragments could be obtained from scFv-phage with a statistically significant correlation. We inferred that the affinity and cross-linking of antibodies had a minimal effect on the relative ranking of antibody internalization in consideration of the diversity of antibodies for both target proteins and binding interactions with the antigens. Moreover, bivalent scFv-Fc did not deviate meaningfully from the monovalent scFv and scFv-phage antibody internalization. Taken together, these results demonstrate that the internalization of phage antibodies can serve as a predictive marker for the internalization of the corresponding antibody.
5. Conclusions
Systematic quantitative and qualitative comparisons of the relative rate or degree of internalization among antibody formats, which could serve as a guide for the evaluation and selection of an antibody candidate for ADCs and FDCs, have not been reported previously. We evaluated antibodies in three different antibody formats (scFv-phage, scFv, and scFv-Fc) in side-by-side comparison studies, using antibodies consisting of diverse characteristics in association with antigens. We showed that the relative trend of antibody internalization can be determined, including the antibodies that may bind to the proteins present in low abundance, as indicated by the low MFI of the antibody associated with the protein prior to endocytosis. Furthermore, we demonstrated that the overall relative trends and kinetics of antibody internalization are retained across scFv-phage, scFv, and scFv-Fc fragments. The statistically significant correlation of scFv-phage antibody internalization with the corresponding scFv and scFv-Fc fragments indicates that the kinetics of scFv-phage internalization can provide insightful information for the assessment of their antibody and antibody fragment internalization prior to the production of the antibody. The results also indicate that the trend of relative internalization can guide an a priori prediction of the “relative ranking order” of internalization from a panel of scFv-phage antibodies. Furthermore, this work demonstrates that the guided selection of the desired endocytic antibody can be rapidly screened and selected directly from the antibody phage library. Additionally, the current approach provides an alternative strategic method for identifying functional internalizing antibodies for the development of ADCs/FDCs and other antibody fragment conjugates.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}