The Hyperoxic-Hypoxic Paradox



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
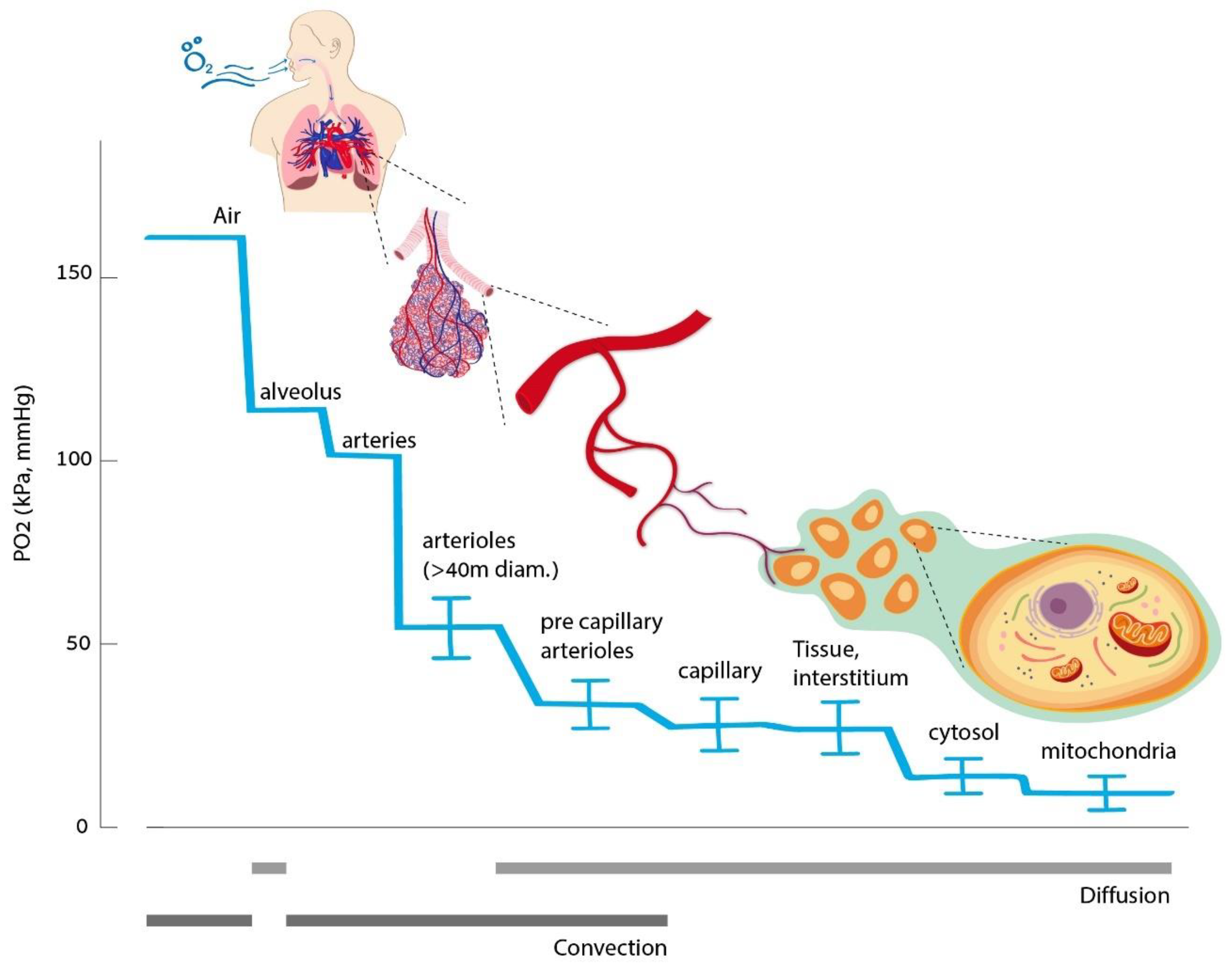
2. Oxygen Homeostasis
3. Hypoxia-Induced Cellular Cascade
3.1. Hypoxic Inducible Factor
3.2. Vascular Endothelial Growth Factor (VEGF)
3.3. Sirtuin
3.4. Mitochondria Biogenesis
3.5. Stem Cells
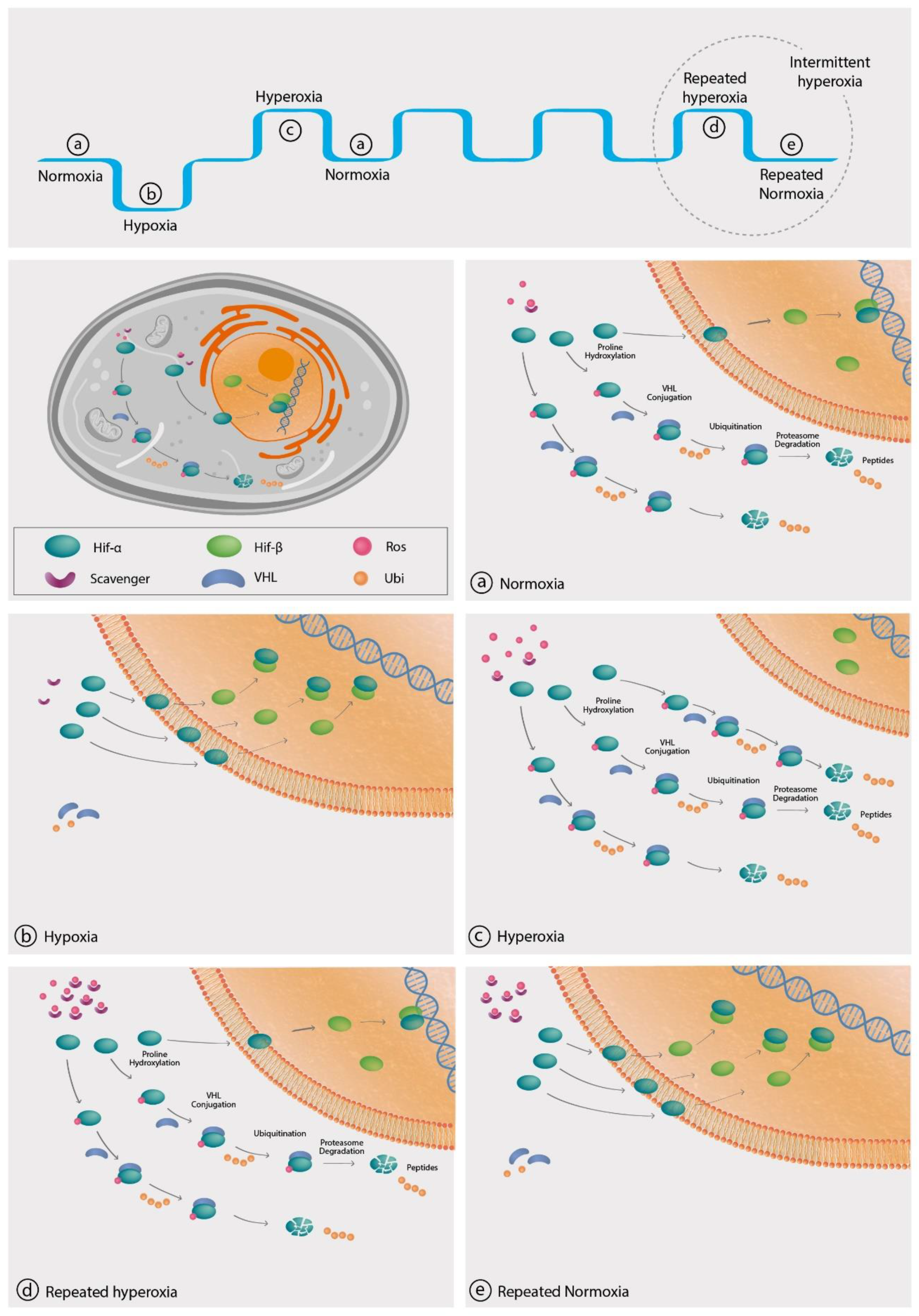
4. The Hyperoxic Hypoxic Paradox
4.1. Hypoxia-Inducible Factor
4.2. VEGF and Angiogenesis
4.3. Sirtuin
4.4. Mitochondria
4.5. Stem Cells
4.6. Oxygen Toxicity
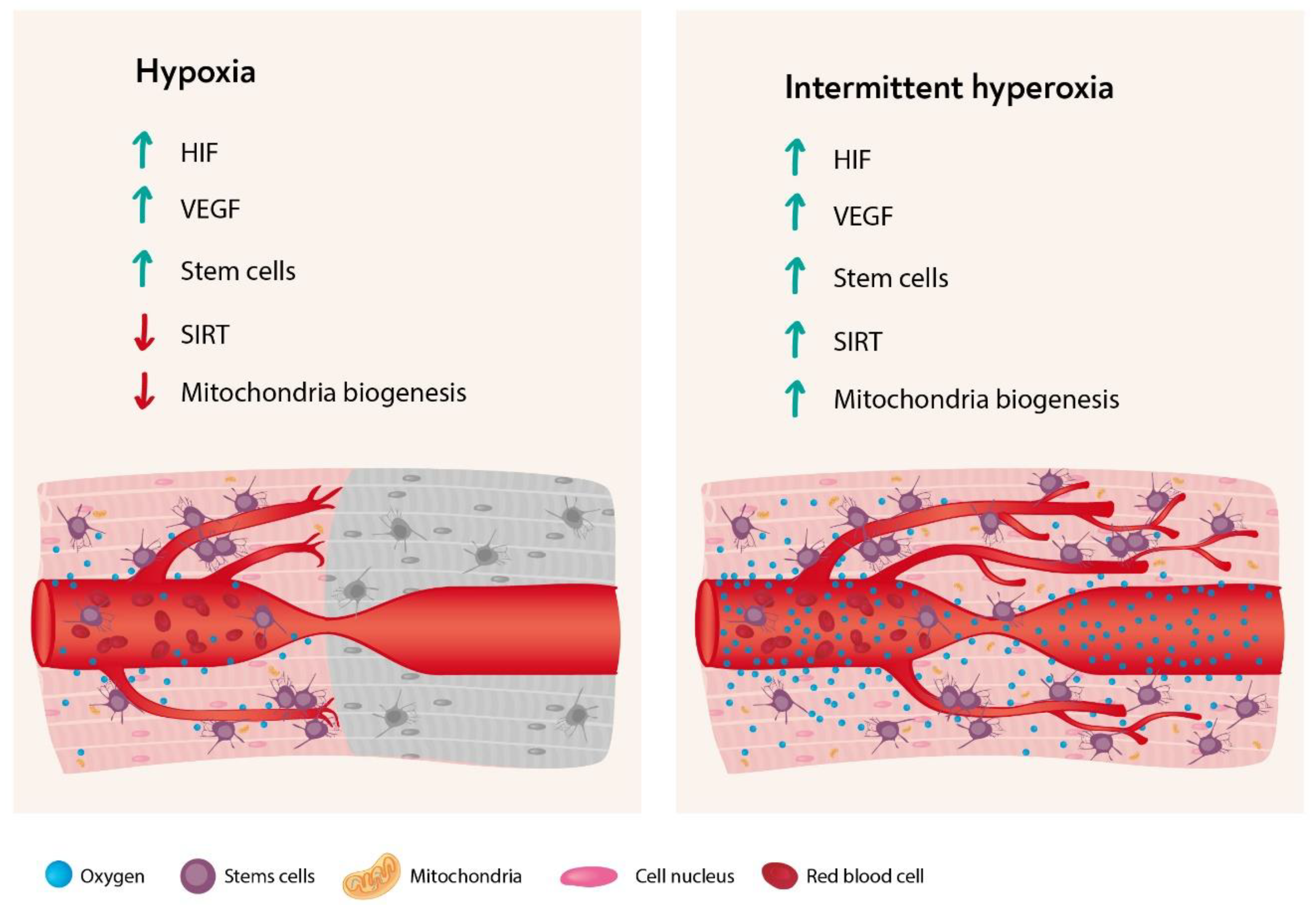
5. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Berner, R.A. Atmospheric oxygen over Phanerozoic time. Proc. Natl. Acad. Sci. USA 1999, 96, 10955–10957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, N.J. Oxygen, animals and oceanic ventilation: An alternative view. Geobiology 2009, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Oxygen-A Critical, but Overlooked, Nutrient. Front. Nutr. 2019, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.A.; Rudenski, A.; Gibson, J.; Howard, L.; O’Driscoll, R. Relating oxygen partial pressure, saturation and content: The haemoglobin-oxygen dissociation curve. Breathe (Sheff) 2015, 11, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Echevarria, M.; Munoz-Cabello, A.M.; Sanchez-Silva, R.; Toledo-Aral, J.J.; Lopez-Barneo, J. Development of cytosolic hypoxia and hypoxia-inducible factor stabilization are facilitated by aquaporin-1 expression. J. Biol. Chem. 2007, 282, 30207–30215. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.P. Oxygen sensors in context. Biochim. Biophys. Acta 2008, 1777, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, U.R.; Fisslthaler, B.; Barbosa-Sicard, E.; Falck, J.R.; Fleming, I.; Busse, R. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J. Cell Sci. 2005, 118, 5489–5498. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, R. Hypoxia and hyperbaric oxygen therapy: A review. Int. J. Gen. Med. 2018, 11, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minet, E.; Ernest, I.; Michel, G.; Roland, I.; Remacle, J.; Raes, M.; Michiels, C. HIF1A gene transcription is dependent on a core promoter sequence encompassing activating and inhibiting sequences located upstream from the transcription initiation site and cis elements located within the 5’UTR. BioChem. Biophys. Res. Commun. 1999, 261, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxic regulation of erythropoiesis and iron metabolism. Am. J. Physiol. Ren. Physiol. 2010, 299, F1–F13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Strehin, I.; Bedelbaeva, K.; Gourevitch, D.; Clark, L.; Leferovich, J.; Messersmith, P.B.; Heber-Katz, E. Drug-induced regeneration in adult mice. Sci. Transl. Med. 2015, 7, 290ra92. [Google Scholar] [CrossRef] [Green Version]
- Adamcio, B.; Sperling, S.; Hagemeyer, N.; Walkinshaw, G.; Ehrenreich, H. Hypoxia inducible factor stabilization leads to lasting improvement of hippocampal memory in healthy mice. Behav. Brain Res. 2010, 208, 80–84. [Google Scholar] [CrossRef]
- Schreiber, T.; Salhofer, L.; Quinting, T.; Fandrey, J. Things get broken: The hypoxia-inducible factor prolyl hydroxylases in ischemic heart disease. Basic Res. Cardiol. 2019, 114, 16. [Google Scholar] [CrossRef]
- Xing, J.; Lu, J. HIF-1alpha Activation Attenuates IL-6 and TNF-alpha Pathways in Hippocampus of Rats Following Transient Global Ischemia. Cell. Physiol. BioChem. 2016, 39, 511–520. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Liang, S.; Lin, B.; Peng, Q.; Zhao, P.; Cui, J.; Rao, Y. Effect of hypoxia-inducible factor-1/vascular endothelial growth factor signaling pathway on spinal cord injury in rats. Exp. Ther. Med. 2017, 13, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Kido, M.; Du, L.; Sullivan, C.C.; Li, X.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J. Am. Coll. Cardiol. 2005, 46, 2116–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkkainen, M.J.; Petrova, T.V. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2000, 19, 5598–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weel, V.; van Tongeren, R.B.; van Hinsbergh, V.W.; van Bockel, J.H.; Quax, P.H. Vascular growth in ischemic limbs: A review of mechanisms and possible therapeutic stimulation. Ann. Vasc. Surg. 2008, 22, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, M.; Hou, T.; Gao, T.; Zhu, W.G.; Yang, Y. Sirtuins in glucose and lipid metabolism. Oncotarget 2017, 8, 1845–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulose, N.; Raju, R. Sirtuin regulation in aging and injury. Biochim. Biophys. Acta 2015, 1852, 2442–2455. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Yao, L.; Ma, X.; Xu, X. Small Molecules as SIRT Modulators. Mini Rev. Med. Chem. 2018, 18, 1151–1157. [Google Scholar] [CrossRef]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1375–H1389. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.N.; Dai, Y.; Zhang, C.H.; Omondi, A.M.; Ghosh, A.; Khanra, I.; Chakraborty, M.; Yu, X.B.; Liang, J. Sirtuins family as a target in endothelial cell dysfunction: Implications for vascular ageing. Biogerontology 2020. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [Green Version]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutierrez-Juarez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008, 8, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firestein, R.; Blander, G.; Michan, S.; Oberdoerffer, P.; Ogino, S.; Campbell, J.; Bhimavarapu, A.; Luikenhuis, S.; de Cabo, R.; Fuchs, C.; et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 2008, 3, e2020. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sadoshima, J. Protection of the heart against ischemia/reperfusion by silent information regulator 1. Trends Cardiovasc. Med. 2011, 21, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 2011, 286, 13869–13878. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, B.; Li, T.; Zhu, Y.; Luo, G.; Jiang, Y.; Tang, F.; Jian, Z.; Xiao, Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. Int. J. Mol. Med. 2018, 41, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Signore, A.P.; Iwai, M.; Cao, G.; Gao, Y.; Chen, J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 2008, 39, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
- Marsin, A.S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gioia, R.; Biella, F.; Citterio, G.; Rizzo, F.; Abati, E.; Nizzardo, M.; Bresolin, N.; Comi, G.P.; Corti, S. Neural Stem Cell Transplantation for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3103. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Peault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, K.; Dakik, P.; Medkour, Y.; Mitrofanova, D.; Titorenko, V.I. Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. Int. J. Mol. Sci. 2019, 20, 2158. [Google Scholar] [CrossRef] [Green Version]
- Alijani, N.; Johari, B.; Moradi, M.; Kadivar, M. A review on transcriptional regulation responses to hypoxia in mesenchymal stem cells. Cell Biol. Int. 2019, 44, 14–26. [Google Scholar] [CrossRef]
- Das, R.; Jahr, H.; van Osch, G.J.; Farrell, E. The role of hypoxia in bone marrow-derived mesenchymal stem cells: Considerations for regenerative medicine approaches. Tissue Eng. Part B Rev. 2010, 16, 159–168. [Google Scholar] [CrossRef]
- Han, Y.; Ren, J.; Bai, Y.; Pei, X.; Han, Y. Exosomes from hypoxia-treated human adipose-derived mesenchymal stem cells enhance angiogenesis through VEGF/VEGF-R. Int. J. Biochem. Cell Biol. 2019, 109, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.H.; Wu, J.; Mou, F.F.; Xie, W.H.; Wang, F.B.; Wang, Q.L.; Fang, J.; Xu, Y.W.; Dong, Y.R.; Liu, J.R.; et al. Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice. FASEB J. 2018, 32, 654–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocco, M.; D’Itri, L.; De Bels, D.; Corazza, F.; Balestra, C. The “normobaric oxygen paradox”: A new tool for the anesthetist? Minerva Anestesiol 2014, 80, 366–372. [Google Scholar] [PubMed]
- Cimino, F.; Balestra, C.; Germonpre, P.; De Bels, D.; Tillmans, F.; Saija, A.; Speciale, A.; Virgili, F. Pulsed high oxygen induces a hypoxic-like response in human umbilical endothelial cells and in humans. J. Appl. Physiol. 2012, 113, 1684–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestra, C.; Germonpre, P. Hypoxia, a multifaceted phenomenon: The example of the “normobaric oxygen paradox”. Eur. J. Appl. Physiol. 2012, 112, 4173–4175. [Google Scholar] [CrossRef]
- Haddad, J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell. Signal. 2002, 14, 879–897. [Google Scholar] [CrossRef]
- Novak, S.; Drenjancevic, I.; Vukovic, R.; Kellermayer, Z.; Cosic, A.; Tolusic Levak, M.; Balogh, P.; Culo, F.; Mihalj, M. Anti-Inflammatory Effects of Hyperbaric Oxygenation during DSS-Induced Colitis in BALB/c Mice Include Changes in Gene Expression of HIF-1alpha, Proinflammatory Cytokines, and Antioxidative Enzymes. Mediat. Inflamm. 2016, 2016, 7141430. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Liang, X.; Chen, D.; Chen, Y.; Doycheva, D.; Tang, J.; Tang, J.; Zhang, J.H. Delayed hyperbaric oxygen therapy promotes neurogenesis through reactive oxygen species/hypoxia-inducible factor-1alpha/beta-catenin pathway in middle cerebral artery occlusion rats. Stroke 2014, 45, 1807–1814. [Google Scholar] [CrossRef]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Hyperbaric oxygen stimulates vasculogenic stem cell growth and differentiation in vivo. J. Appl. Physiol. 2009, 106, 711–728. [Google Scholar] [CrossRef]
- Yu, Q.H.; Zhang, P.X.; Liu, Y.; Liu, W.; Yin, N. Hyperbaric oxygen preconditioning protects the lung against acute pancreatitis induced injury via attenuating inflammation and oxidative stress in a nitric oxide dependent manner. BioChem. Biophys. Res. Commun. 2016, 478, 93–100. [Google Scholar] [CrossRef]
- Ren, P.; Kang, Z.; Gu, G.; Liu, Y.; Xu, W.; Tao, H.; Zhang, J.H.; Sun, X.; Ji, H. Hyperbaric oxygen preconditioning promotes angiogenesis in rat liver after partial hepatectomy. Life Sci. 2008, 83, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Salhanick, S.D.; Belikoff, B.; Orlow, D.; Holt, D.; Reenstra, W.; Buras, J.A. Hyperbaric oxygen reduces acetaminophen toxicity and increases HIF-1alpha expression. Acad. Emerg. Med. 2006, 13, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Ren, P.; Kang, Z.; Du, J.; Lian, Q.; Liu, Y.; Zhang, J.H.; Sun, X. Up-regulated HIF-1alpha is involved in the hypoxic tolerance induced by hyperbaric oxygen preconditioning. Brain Res. 2008, 1212, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.; Adler, I.; Manaenko, A.; Schwarz, S.C.; Walkinshaw, G.; Arend, M.; Flippin, L.A.; Storch, A.; Schwarz, J. Non-hypoxic stabilization of hypoxia-inducible factor alpha (HIF-alpha): Relevance in neural progenitor/stem cells. Neurotox. Res. 2009, 15, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Cahill, J.; Yamaguchi-Okada, M.; Zhang, J.H. Oxygen treatment after experimental hypoxia-ischemia in neonatal rats alters the expression of HIF-1alpha and its downstream target genes. J. Appl. Physiol. 2006, 101, 853–865. [Google Scholar] [CrossRef]
- Bai, X.; Sun, B.; Pan, S.; Jiang, H.; Wang, F.; Krissansen, G.W.; Sun, X. Down-regulation of hypoxia-inducible factor-1alpha by hyperbaric oxygen attenuates the severity of acute pancreatitis in rats. Pancreas 2009, 38, 515–522. [Google Scholar] [CrossRef]
- Li, Z.; Li, M.; Li, X.; Zhang, M.; Zhao, Y.; Ren, W.; Cheng, J.; Wang, X. Hyperbaric oxygen inhibits venous neointimal hyperplasia following arteriovenous fistulization. Int. J. Mol. Med. 2017, 39, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chang, Q.; Cox, R.A.; Gong, X.; Gould, L.J. Hyperbaric oxygen attenuates apoptosis and decreases inflammation in an ischemic wound model. J. Investig. Dermatol. 2008, 128, 2102–2112. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Marti, H.H.; Veltkamp, R. Hyperbaric oxygen reduces tissue hypoxia and hypoxia-inducible factor-1 alpha expression in focal cerebral ischemia. Stroke 2008, 39, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, C.; Calvert, J.W.; Colohan, A.R.; Zhang, J.H. Multiple effects of hyperbaric oxygen on the expression of HIF-1 alpha and apoptotic genes in a global ischemia-hypotension rat model. Exp. Neurol. 2005, 191, 198–210. [Google Scholar] [CrossRef]
- Duan, S.; Shao, G.; Yu, L.; Ren, C. Angiogenesis contributes to the neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Int. J. Neurosci. 2015, 125, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.J.; Li, Y.P.; Peng, Z.Y.; Xu, J.J.; Kang, Z.M.; Xu, W.G.; Tao, H.Y.; Ostrowski, R.P.; Zhang, J.H.; Sun, X.J. Mechanism of ischemic tolerance induced by hyperbaric oxygen preconditioning involves upregulation of hypoxia-inducible factor-1alpha and erythropoietin in rats. J. Appl. Physiol. 2008, 104, 1185–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.Y.; Sung, P.H.; Chung, S.Y.; Hsu, S.L.; Chung, W.J.; Sheu, J.J.; Hsueh, S.K.; Chen, K.H.; Wu, R.W.; Yip, H.K. Hyperbaric Oxygen Therapy Enhanced Circulating Levels of Endothelial Progenitor Cells and Angiogenesis Biomarkers, Blood Flow, in Ischemic Areas in Patients with Peripheral Arterial Occlusive Disease. J. Clin Med. 2018, 7, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano-Hernandez, Y.M.; Contreras-Mendez, L.; de Los Angeles Hernandez-Cueto, M.; Muand Oz-Medina, J.E.; Santillan-Verde, M.A.; Barbosa-Cabrera, R.E.; Delgado-Quintana, C.A.; Trejo-Rosas, S.; Santacruz-Tinoco, C.E.; Gonzalez-Ibarra, J.; et al. Modification of HIF-1alpha, NF-akappaB, IGFBP-3, VEGF and adiponectin in diabetic foot ulcers treated with hyperbaric oxygen. Undersea Hyperb. Med. 2019, 46, 35–44. [Google Scholar]
- Hsu, S.L.; Yin, T.C.; Shao, P.L.; Chen, K.H.; Wu, R.W.; Chen, C.C.; Lin, P.Y.; Chung, S.Y.; Sheu, J.J.; Sung, P.H.; et al. Hyperbaric oxygen facilitates the effect of endothelial progenitor cell therapy on improving outcome of rat critical limb ischemia. Am. J. Transl. Res. 2019, 11, 1948–1964. [Google Scholar]
- Sureda, A.; Batle, J.M.; Martorell, M.; Capo, X.; Tejada, S.; Tur, J.A.; Pons, A. Antioxidant Response of Chronic Wounds to Hyperbaric Oxygen Therapy. PLoS ONE 2016, 11, e0163371. [Google Scholar] [CrossRef] [Green Version]
- Hadanny, A.; Efrati, S. Oxygen—A limiting factor for brain recovery. Crit. Care 2015, 19, 307. [Google Scholar] [CrossRef] [Green Version]
- Vadas, D.; Kalichman, L.; Hadanny, A.; Efrati, S. Hyperbaric Oxygen Environment Can Enhance Brain Activity and Multitasking Performance. Front. Integr. Neurosci. 2017, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Efrati, S.; Ben-Jacob, E. Reflections on the neurotherapeutic effects of hyperbaric oxygen. Expert Rev. Neurother. 2014, 14, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Tal, S.; Hadanny, A.; Berkovitz, N.; Sasson, E.; Ben-Jacob, E.; Efrati, S. Hyperbaric oxygen may induce angiogenesis in patients suffering from prolonged post-concussion syndrome due to traumatic brain injury. Restor. Neurol. Neurosci. 2015, 33, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Tal, S.; Hadanny, A.; Sasson, E.; Suzin, G.; Efrati, S. Hyperbaric Oxygen Therapy Can Induce Angiogenesis and Regeneration of Nerve Fibers in Traumatic Brain Injury Patients. Front. Hum. Neurosci. 2017, 11, 508. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Chung, S.; Hwang, J.W.; Rajendrasozhan, S.; Sundar, I.K.; Dean, D.A.; McBurney, M.W.; Guarente, L.; Gu, W.; Ronty, M.; et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Investig. 2012, 122, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Fang, Z.; Yang, Q.; Dong, H.; Lu, Y.; Lei, C.; Xiong, L. SirT1 mediates hyperbaric oxygen preconditioning-induced ischemic tolerance in rat brain. J. Cereb. Blood Flow Metab. 2013, 33, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, F.; Huang, J.W.; Ding, P.Y.; Zang, H.G.; Kou, Z.J.; Li, T.; Fan, J.; Peng, Z.W.; Yan, W.J. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav. Brain Res. 2016, 309, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Manaenko, A.; Bian, H.; Guo, Z.; Huang, J.L.; Guo, Z.N.; Yang, P.; Tang, J.; Zhang, J.H. Hyperbaric Oxygen Reduces Infarction Volume and Hemorrhagic Transformation Through ATP/NAD(+)/Sirt1 Pathway in Hyperglycemic Middle Cerebral Artery Occlusion Rats. Stroke 2017, 48, 1655–1664. [Google Scholar] [CrossRef]
- Hong-Qiang, H.; Mang-Qiao, S.; Fen, X.; Shan-Shan, L.; Hui-Juan, C.; Wu-Gang, H.; Wen-Jun, Y.; Zheng-Wu, P. Sirt1 mediates improvement of isoflurane-induced memory impairment following hyperbaric oxygen preconditioning in middle-aged mice. Physiol. Behav. 2018, 195, 1–8. [Google Scholar] [CrossRef]
- Suzuki, J. Endurance performance is enhanced by intermittent hyperbaric exposure via up-regulation of proteins involved in mitochondrial biogenesis in mice. Physiol. Rep. 2017, 5, e13349. [Google Scholar] [CrossRef]
- DeCato, T.W.; Bradley, S.M.; Wilson, E.L.; Harlan, N.P.; Villela, M.A.; Weaver, L.K.; Hegewald, M.J. Effects of sprInt. interval training on cardiorespiratory fitness while in a hyperbaric oxygen environment. Undersea Hyperb. Med. 2019, 46, 117–124. [Google Scholar]
- Chavko, M.; Harabin, A.L. Regional lipid peroxidation and protein oxidation in rat brain after hyperbaric oxygen exposure. Free Radic. Biol. Med. 1996, 20, 973–978. [Google Scholar] [CrossRef]
- Gutsaeva, D.R.; Suliman, H.B.; Carraway, M.S.; Demchenko, I.T.; Piantadosi, C.A. Oxygen-induced mitochondrial biogenesis in the rat hippocampus. Neuroscience 2006, 137, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Lippert, T.; Borlongan, C.V. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. Ther. 2019, 25, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.B.; Ren, H.; Zhao, H.; Chi, Y.; Chen, K.; Zhou, B.; Liu, Y.J.; Zhang, L.; Xu, B.; Liu, B.; et al. Hypoxia-inducible factor (HIF)-1 alpha directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF). Carcinogenesis 2008, 29, 1853–1861. [Google Scholar] [CrossRef]
- Shandley, S.; Wolf, E.G.; Schubert-Kappan, C.M.; Baugh, L.M.; Richards, M.F.; Prye, J.; Arizpe, H.M.; Kalns, J. Increased circulating stem cells and better cognitive performance in traumatic brain injury subjects following hyperbaric oxygen therapy. Undersea Hyperb. Med. 2017, 44, 257–269. [Google Scholar] [CrossRef]
- Heyboer, M., 3rd; Milovanova, T.N.; Wojcik, S.; Grant, W.; Chin, M.; Hardy, K.R.; Lambert, D.S.; Logue, C.; Thom, S.R. CD34+/CD45-dim stem cell mobilization by hyperbaric oxygen—Changes with oxygen dosage. Stem Cell Res. 2014, 12, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Thom, S.R.; Bhopale, V.M.; Velazquez, O.C.; Goldstein, L.J.; Thom, L.H.; Buerk, D.G. Stem cell mobilization by hyperbaric oxygen. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1378–H1386. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Yang, Y.J.; Xie, M.; Yu, X.H.; Liu, C.T.; Wang, X. Proliferation of neural stem cells correlates with Wnt-3 protein in hypoxic-ischemic neonate rats after hyperbaric oxygen therapy. Neuroreport 2007, 18, 1753–1756. [Google Scholar] [CrossRef]
- Feng, Z.; Liu, J.; Ju, R. Hyperbaric oxygen treatment promotes neural stem cell proliferation in the subventricular zone of neonatal rats with hypoxic-ischemic brain damage. Neural Regen. Res. 2013, 8, 1220–1227. [Google Scholar]
- Yang, Y.; Wei, H.; Zhou, X.; Zhang, F.; Wang, C. Hyperbaric oxygen promotes neural stem cell proliferation by activating vascular endothelial growth factor/extracellular signal-regulated kinase signaling after traumatic brain injury. Neuroreport 2017, 28, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yang, Q.W.; Wang, S.N.; Wang, J.Z.; Wang, Q.; Wang, Y.; Luo, Y.J. Hyperbaric oxygen therapy improves neurogenesis and brain blood supply in piriform cortex in rats with vascular dementia. Brain Inj. 2010, 24, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Wang, X.L.; Yu, X.H.; Wang, X.; Xie, M.; Liu, C.T. Hyperbaric oxygen induces endogenous neural stem cells to proliferate and differentiate in hypoxic-ischemic brain damage in neonatal rats. Undersea Hyperb. Med. 2008, 35, 113–129. [Google Scholar] [PubMed]
- Lee, Y.S.; Chio, C.C.; Chang, C.P.; Wang, L.C.; Chiang, P.M.; Niu, K.C.; Tsai, K.J. Long course hyperbaric oxygen stimulates neurogenesis and attenuates inflammation after ischemic stroke. Mediat. Inflamm. 2013, 2013, 512978. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Wang, J.; Cao, Y.; Ren, Q.; Zhao, L.; Li, X.; Wang, J. Hyperbaric oxygenation promotes neural stem cell proliferation and protects the learning and memory ability in neonatal hypoxic-ischemic brain damage. Int. J. Clin. Exp. Pathol. 2015, 8, 1752–1759. [Google Scholar]
- Thom, S.R.; Milovanova, T.N.; Yang, M.; Bhopale, V.M.; Sorokina, E.M.; Uzun, G.; Malay, D.S.; Troiano, M.A.; Hardy, K.R.; Lambert, D.S.; et al. Vasculogenic stem cell mobilization and wound recruitment in diabetic patients: Increased cell number and intracellular regulatory protein content associated with hyperbaric oxygen therapy. Wound Repair Regen. 2011, 19, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Bekheit, M.; Baddour, N.; Katri, K.; Taher, Y.; El Tobgy, K.; Mousa, E. Hyperbaric oxygen therapy stimulates colonic stem cells and induces mucosal healing in patients with refractory ulcerative colitis: A prospective case series. BMJ Open Gastroenterol. 2016, 3, e000082. [Google Scholar] [CrossRef]
- Gardin, C.; Bosco, G.; Ferroni, L.; Quartesan, S.; Rizzato, A.; Tatullo, M.; Zavan, B. Hyperbaric Oxygen Therapy Improves the Osteogenic and Vasculogenic Properties of Mesenchymal Stem Cells in the Presence of Inflammation In Vitro. Int. J. Mol. Sci. 2020, 21, 1452. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.S.; Ueng, S.W.; Niu, C.C.; Yuan, L.J.; Yang, C.Y.; Chen, W.J.; Lee, M.S.; Chen, J.K. Effects of hyperbaric oxygen on the osteogenic differentiation of mesenchymal stem cells. BMC Musculoskelet. Disord. 2014, 15, 56. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.H.; Chang, S.S.; Chang, G.J.; Chen, A.C.; Cheng, C.Y.; Chen, S.C.; Chan, Y.S. The Effect of Hyperbaric Oxygen Treatment on Myoblasts and Muscles After Contusion Injury. J. Orthop. Res. 2020, 38, 329–335. [Google Scholar] [CrossRef]
- Chaillou, T.; Lanner, J.T. Regulation of myogenesis and skeletal muscle regeneration: Effects of oxygen levels on satellite cell activity. FASEB J. 2016, 30, 3929–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Enomoto, M.; Shimoda, M.; Okawa, A.; Miyakawa, S.; Yagishita, K. Enhancement of satellite cell differentiation and functional recovery in injured skeletal muscle by hyperbaric oxygen treatment. J. Appl. Physiol. 2014, 116, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadori, A.; Agoston, V.A.; Demeter, K.; Hadinger, N.; Varady, L.; Kohidi, T.; Gobl, A.; Nagy, Z.; Madarasz, E. Survival and differentiation of neuroectodermal cells with stem cell properties at different oxygen levels. Exp. Neurol. 2011, 227, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Liu, Z.G.; Liu, X.J.; Chen, Q.X. Umbilical cord-derived mesenchymal stem cell transplantation combined with hyperbaric oxygen treatment for repair of traumatic brain injury. Neural Regen. Res. 2016, 11, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.K.; Cao, H.H.; Ying, X.; Yu, H.L. Effect of mesenchymal stem cells transplantation combining with hyperbaric oxygen therapy on rehabilitation of rat spinal cord injury. Asian Pac. J. Trop. Med. 2015, 8, 468–473. [Google Scholar] [CrossRef]
- Pan, H.C.; Chin, C.S.; Yang, D.Y.; Ho, S.P.; Chen, C.J.; Hwang, S.M.; Chang, M.H.; Cheng, F.C. Human amniotic fluid mesenchymal stem cells in combination with hyperbaric oxygen augment peripheral nerve regeneration. Neurochem. Res. 2009, 34, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Meduru, S.; Pandian, R.P.; Rivera, B.K.; Kuppusamy, P. Effect of oxygenation on stem-cell therapy for myocardial infarction. Adv. Exp. Med. Biol. 2011, 701, 175–181. [Google Scholar]
- Khan, M.; Meduru, S.; Mohan, I.K.; Kuppusamy, M.L.; Wisel, S.; Kulkarni, A.; Rivera, B.K.; Hamlin, R.L.; Kuppusamy, P. Hyperbaric oxygenation enhances transplanted cell graft and functional recovery in the infarct heart. J. Mol. Cell. Cardiol. 2009, 47, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Pena-Villalobos, I.; Casanova-Maldonado, I.; Lois, P.; Prieto, C.; Pizarro, C.; Lattus, J.; Osorio, G.; Palma, V. Hyperbaric Oxygen Increases Stem Cell Proliferation, Angiogenesis and Wound-Healing Ability of WJ-MSCs in Diabetic Mice. Front. Physiol. 2018, 9, 995. [Google Scholar] [CrossRef] [Green Version]
- Torbati, D.; Church, D.F.; Keller, J.M.; Pryor, W.A. Free radical generation in the brain precedes hyperbaric oxygen-induced convulsions. Free Radic. Biol. Med. 1992, 13, 101–106. [Google Scholar] [CrossRef]
- Kellogg, R.H. “La Pression barometrique”: Paul Bert’s hypoxia theory and its critics. Respir. Physiol. 1978, 34, 1–28. [Google Scholar] [CrossRef]
- Leach, R.M.; Rees, P.J.; Wilmshurst, P. Hyperbaric oxygen therapy. BMJ 1998, 317, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K.; Torbati, D.; Tao, H.Y.; Ni, G.T. Oxygen Toxicity. In Textbook of Hyperbaric Medicine, 4th ed.; Best Publishing Company: North Palm Beach, FL, USA, 1999; pp. 47–59. [Google Scholar]
- Hadanny, A.; Meir, O.; Bechor, Y.; Fishlev, G.; Bergan, J.; Efrati, S. The safety of hyperbaric oxygen treatment--retrospective analysis in 2334 patients. Undersea Hyperb. Med. 2016, 43, 113–122. [Google Scholar] [PubMed]
- Hadanny, A.; Meir, O.; Bechor, Y.; Fishlev, G.; Bergan, J.; Efrati, S. Seizures during hyperbaric oxygen therapy: Retrospective analysis of 62,614 treatment sessions. Undersea Hyperb. Med. 2016, 43, 21–28. [Google Scholar] [PubMed]
- Hadanny, A.; Zubari, T.; Tamir-Adler, L.; Bechor, Y.; Fishlev, G.; Lang, E.; Polak, N.; Bergan, J.; Friedman, M.; Efrati, S. Hyperbaric oxygen therapy effects on pulmonary functions: A prospective cohort study. BMC Pulm. Med. 2019, 19, 148. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadanny, A.; Efrati, S. The Hyperoxic-Hypoxic Paradox. Biomolecules 2020, 10, 958. https://doi.org/10.3390/biom10060958
Hadanny A, Efrati S. The Hyperoxic-Hypoxic Paradox. Biomolecules. 2020; 10(6):958. https://doi.org/10.3390/biom10060958
Chicago/Turabian StyleHadanny, Amir, and Shai Efrati. 2020. "The Hyperoxic-Hypoxic Paradox" Biomolecules 10, no. 6: 958. https://doi.org/10.3390/biom10060958
APA StyleHadanny, A., & Efrati, S. (2020). The Hyperoxic-Hypoxic Paradox. Biomolecules, 10(6), 958. https://doi.org/10.3390/biom10060958