1. Introduction
Fluorinated compounds have remained important in the field of lead compound discovery due to their wide therapeutic and agricultural applications [
1]. The unmet need for a fluorine-incorporated lead compound presents an unremitting impetus for the development of selectively efficient fluorination methods. Compared to the vast amount of synthetic fluorinated compounds, naturally occurring fluorinated natural products are extremely rare, most of which are fluorinated fatty acids with different chain lengths discovered in sub-tropical and tropical plants [
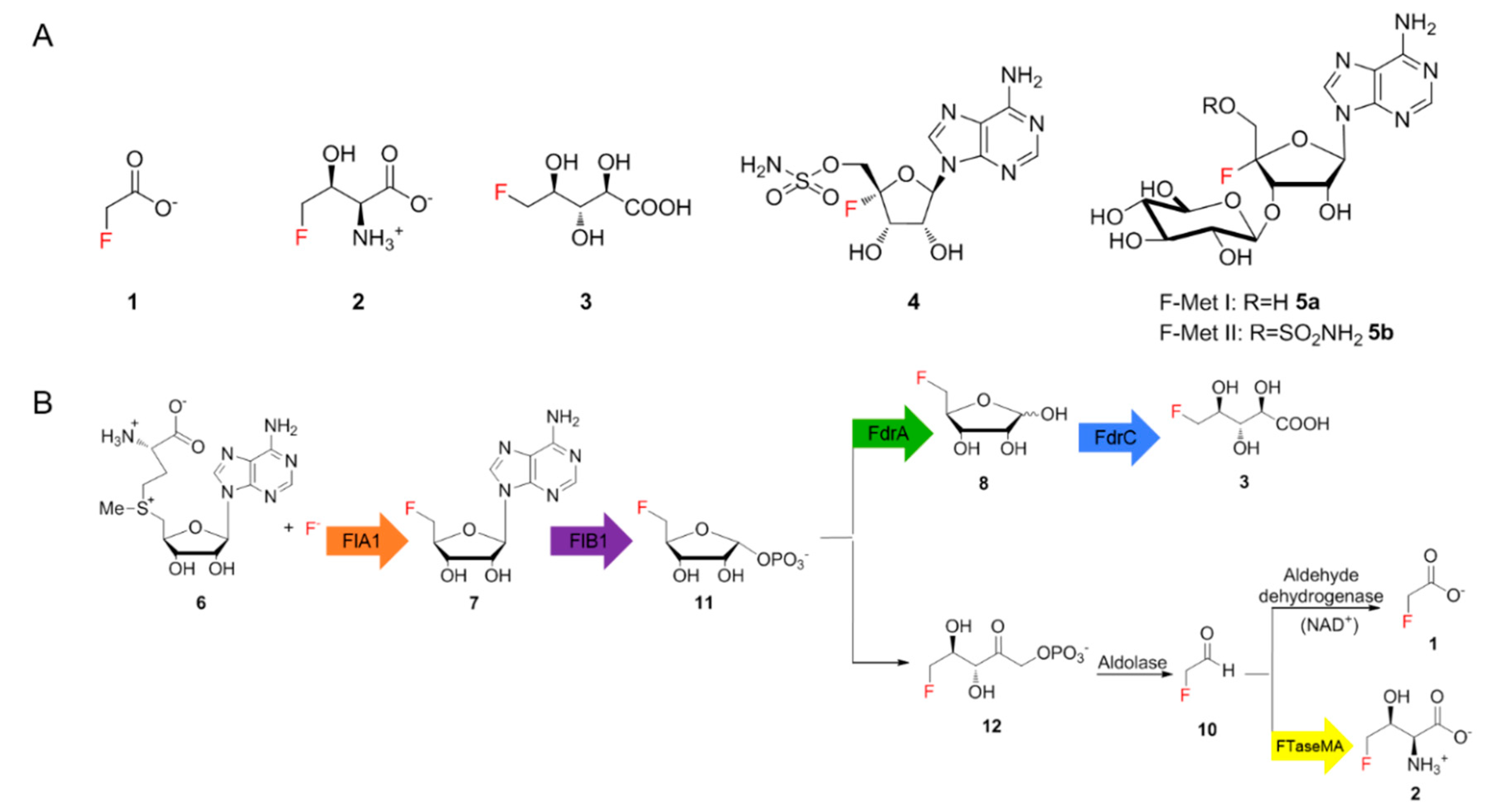
1]. Fewer than thirty fluorinated natural products have been identified so far, including six structurally different ones of bacterial origin, such as fluoroacetate (FAc)
1, 4-fluorothreonine (4-FT)
2 [1], 5-fluoro-2,3,4-trihydroxypentanoic acid (FHPA)
3 [
2], nucleocidin
4 [
3] and two new glycosylated nucleocidin derivatives (
5a,
5b) (
Figure 1A) [
4]. Of these,
1 is a toxin that inhibits the citric acid cycle while
2 and
4 were discovered as antimicrobial metabolites. The biological function of
3 in the producing strain remains elusive. The scarcity of fluorinated compounds found in nature, compared to thousands of brominated or iodinated compounds, mainly results from the high electronegativity of fluorine, which precludes the common strategy for halogenation that involves the oxidation of fluoride ions [
5]. In addition, the solvation tendency of fluoride ions in aqueous solutions causes fluoride to need extra energy to become a good nucleophile. Such desolvation processes elevate the difficulty of biochemically incorporating fluorine into complex organic molecules [
5].
For decades,
Streptomyces cattleya remained the only genetically tractable strain producing two fluorinated natural products,
1 and
2. The major breakthrough was the identification of the first fluorination enzyme, fluorinase, which converts
S-adenosyl-l-methionine (SAM)
6 and fluoride ions to generate 5′-fluoro-5′-deoxy-adenosine (5′-FDA
7) [
6,
7]. Subsequent studies resulted in the elucidation of the biosynthetic pathway of
1 and
2 in
S. cattleya as illustrated in
Figure 1B [
8]. Recent studies demonstrated that two genes,
fthB and
fthC, are highly conserved in the biosynthetic gene clusters (BGCs) that direct the production of
2 [
9]. While the genetic inactivation of
fthB had no perturbation in secreted
2 in the
S. cattleya variant as observed in
19F-NMR analysis, biochemical analysis indicated that FthB, an aminoacyl RNA deacylase, plays an essential role in the detoxification of
2, by counteracting the misacylation of fluorothreonyl-tRNA which otherwise would be misincorporated into protein in place of L-threonine [
9]. While knocking out
fthC significantly reduced the concentration of
2 in culture broth [
9], the intracellular
2 was accumulated in the
S. cattleya variant, indicating that FthC is a 4-FT exporter. The efforts towards understanding the fluorination enzyme, fluorometabolisms in
S. cattleya and related biotransformations have offered a new bio-based approach to generating high-value fluorinated chemicals [
10].
In the last decade, advanced genome sequencing technologies have enabled the use of genome mining strategies to identify several other potential fluorometabolite-producing strains [
2,
11]. Recent studies revealed that
Streptomyces sp. MA37 (MA37), a soil isolate, is a talented natural product producing strain [
12,
13,
14,
15]. In particular, MA37 produces
1,
2 and a series of unidentified fluorinated metabolites as observed in
19F-NMR analysis of the supernatant of the culture broth of MA37 [
11]. It has been shown that
1 and
2 in MA37 originate from the same biosynthetic pathway as the one in
S. cattleya [
11,
16]. A combination of chemical synthesis, bioinformatic analysis and biochemical assays allowed the discovery of
3 as a new fluorometabolite and the identification of the second
fdr biosynthetic gene cluster (BGC) in MA37 [
2]. However, the rest of the fluorometabolites observed in MA37 remain to be determined.
Here we describe the identification of a naturally occurring fluorometabolite, 5-fluoro-5-deoxy-D-ribulose (5-FDRul) 9, from the culture broth of MA37, using a combination of genetic inactivation, chemo-enzymatic synthesis and 19F-NMR comparison. Although reported before as chemical probes during the previous fluorometabolism study in S. cattleya, this is the first report indicating that 9 is a naturally occurring fluorinated compound. During the course of our studies, we also observed that the production of several fluorinated natural products was upregulated among several MA37 fluorometabolite biosynthesis related gene inactivation variants as observed in 19F-NMR analysis, suggesting that the inactivation of genes responsible for downstream fluorometabolite biosynthesis may influence metabolic reflux and direct accumulated biosynthetic intermediates to other previously unnoticed pathways in MA37.
2. Experimental Section
2.1. Fermentation Conditions
E. coli strains were grown in Luria-Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl) or LB agar (1.5% agar) at 37 °C, supplemented with the corresponding antibiotics. E. coli DH10B was used as the routine cloning strain for DNA manipulations. E. coli ET12567 (pUZ8002), a DNA methylation deficiency stain, served as conjugal donor. MA37 was grown in ISP2 medium and supplemented with apramycin (20 μM) if needed. For mycelia generation, MA37 was inoculated from an ISP2 agar palate to YEME liquid medium (50 mL) and harvested after 2 d shake incubation (180 rpm, 28 °C). For fluorometabolite production, MA37 wild type and the confirmed in-frame deletion variants were grown on ISP2 agar medium for 4 d before being inoculated into ISP2 liquid medium supplemented with KF (2.5 mM). The seed culture was shake-incubated for 2 d (180 rpm, 28 °C). Seed culture (1 mL) was then inoculated into the same ISP2 medium with KF (50 mL) and shake-incubated for 10–14 d. The supernatant of the culture was obtained by removing the mycelium with a centrifugation step (4600 rpm, 20 min) and freeze-dried. The crude extract was supplemented with D2O and subjected to 19F-NMR analysis after 10–14 d fermentation.
2.2. Genomic DNA Isolation
The genomic DNA of MA37 in this study was extracted from 2 mL cell culture. Cell pellet was harvested after 3 d culture in ISP2 medium by centrifugation and resuspended in 500 μL SET buffer (100 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0). The cell suspension was mixed with lysozyme (4 mg/mL, final concentration) and incubated at 37 °C for 30 min. SDS (60 μL, 10% (w/v)) and NaCl (200 μL, 5 M) were then added to the mixture, followed by further incubation at 60 °C for 30 min. The protein was precipitated with a mixture of phenol, chloroform and isoamylol (500 μL, ratio of 25:24:1) and the resultant mixture was mixed by vortex. The water fraction was obtained after a centrifugation step and transferred to a new Eppendorf tube with isopropanol for DNA precipitation (0.8 volumes). The precipitated DNA was washed with 75% (v/v) ethanol, followed by a second wash with 100% ethanol. The DNA pellet was dried at room temperature and dissolved in sterile MiliQ water (200 μL).
2.3. The Construction of Plasmids Used for Gene Deletion
To generate the deletion vector for in-frame deletion in MA37, two homologous arms were amplified by PCR using MA37 genomic DNA as a template (fthCMA left arm forward: ggc cag tgc caa gct tGG AAT GAA CCC CCA GGA GAC CCG CG, reverse: gcg cag gat acc cgg ACG GTC GGC CGG CAC TTC GAG GGG; right arm forward: CGG GTG ATC CTG CGC AGG GTC GGG GCG G, reverse: aca tga tta cga att cTG CCG GAT CCG CAC GGC CAC GGA GC; fthBMA left arm forward: ggc cag tgc caa gct tCG CGA AGG GCC CGC GTC CGG TAC AC, reverse: cac gga aat cac cga GCC CTA CCG CTG CCC ATG GTG TTC G; right arm forward: TCG GTG ATT TCC GTG CTC TCC GTG G, reverse: aca tga tta cga att cTC TCG TTC GCG GTC AGA TGG AGG AC; fdrA left arm forward: ggc cag tgc caa gct tGC AGG GCG GTG GCG CGT CCG ATG CCG CG, reverse: tcc ccg ttc gcg cag GCA TGT CCT CGA TCT TGC GT; right arm forward: CTG CGC GAA CGG GGA AAC CCC TTC, reverse: gcg cgg ccg cgg atc cCG CCA CCG GGC TGA TGG CCC TG; fdrB left arm forward: ggc cag tgc caa gct tTC TGG TGC CCG ACG GTG ACC GCG AC, reverse: gca atc tca acc acc GCC TGA AGG AAT GGG AGG CTC CGC C; right arm forward: GGT GGT TGA GAT TGC ACG GCA T, reverse: gcg cgg ccg cgg atc cAG CGC GCC CAG TTC GAG AAC CCC). The homologous arms were ligated to the linearized (HindIII, EcoRI for fthBMA and fthCMA deletion and BamHI, HindIII for the rest) temperature-sensitive E. coli–Streptomyces shuttle vector pKC1139 via one pot in-fusion cloning (TAKARA) according to the protocol of the manufacturer (Clontech, TaKaRa, Shiga, Japan). The correct deletion construct was screened by PCR and confirmed by DNA sequencing.
2.4. The E. coli–Streptomyces Conjugation and Double-Crossover Variant Generation
The confirmed deletion construct was introduced to E. coli ET12567 for the E. coli–Streptomyces conjugation. As MA37 is bold and produces no spores, mycelia were used for conjugation instead for spores, and were harvested in YEME culture after 2 d shake incubation. The E. coli donor strain (2 mL) was harvested and washed with fresh LB medium (3×, 1.5 mL), before being mixed with MA37 mycelia (200 μL) and spread onto an MS agar plate (Mannitol 2%, Soya bean flour 2%, agar 2%, pH 7.5). The plate was cultured overnight (28 °C, 12–16 h) and overlaid with apramycin and nalidixic acid (50 μM, final concentration). The exoconjugates were purified and single-crossover variants were isolated by incubation at 37 °C to promote plasmid loss. Double-crossover mutant was then acquired by multiple steps of subculture onto an antibiotic-free ISP2 agar plate and confirmed by PCR screening and genomic DNA sequencing.
2.5. The Synthesis of 5-FDR
The 5-fluoro-5-deoxy-D-ribose (5-FDR)
8 synthesis protocol was taken from the literature [
17,
18]. D-(-)-ribose
14 (1.5 g, 1 eq) was added to a round-bottom flask (250 mL) with acetone (80 mL) and cooled under an ice bath. Then, 2,2-dimethoxypropane (2.6 g, 2.5 eq) was added into the mixture. Perchloric acid was added dropwise into the mixture for 5 min. The ice bath was removed and the reaction mixture stirred at room temperature overnight. The reaction mixture turned orange the next morning. Methanol (2.02 mL, 5 eq) was added into the mixture and it was stirred for 3 h. The reaction was quenched by the saturated addition of sodium hydrogen carbonate. The suspension was filtered and rotary evaporated to minimum volume (aqueous phase). The aqueous phase was extracted by ethyl acetate (3 × 100 mL, organic phase). The combined organic phase was dried by anhydrous magnesium sulphate, filtered and dried under vacuum. Then, 1-methoxy-2,2-isopropyliden-α/β-D-ribofuranose 15 (1 g, 1 equiv) was oven-dried and placed under an argon atmosphere system. Dry pyridine (3 mL) was added into the system and the mixture was cooled under an ice bath. After 5 min,
p-toluenesulfonyl chloride (1.5 equiv) was added into the mixture, which was stirred under the ice bath for 2 h. When the reaction completed, the mixture was poured into 5 mL of ice-cold water with vigorous stirring, and the precipitate formed. The precipitate was filtered, washed by ice-cold water (5 × 5 mL) and dried under a freeze drier. Then, 2,3-O-isopropylidene-5-O-(
p-toluenesulfonyl)-β,D-ribofuranoside 16 (600 mg, 1.68 mmol, 1 equiv) was added into a flame-dried round-bottom flask and connected to the reflux system. The argon cycle was applied to the system. Anhydrous acetonitrile (7 mL) was added, followed by
tetra-
n-butylammonium fluoride (TBAF) (2 mL). The system was heated to 80 °C under reflux overnight. The reaction mixture was dried under vacuum the following morning. The dried product was purified by a silica gel column (mobile phase: petroleum ether:ethyl acetate 10:1). Aqueous sulphuric acid (0.2 M) was mixed into the purified product and heated overnight. The reaction mixture was cooled to room temperature the following morning and neutralized by barium carbonate. The suspension was centrifuged and the supernatant was freeze-dried.
2.6. The Generation of 5-FDRul
For the preparation of 5-FDRul 9, the synthesized 5-FDR 8 (1 mM) was incubated with immobilized glucose isomerase (30 mg) in KH2PO4 buffer (50 mM, pH 6.8) at 37 °C for 6 h. The enzyme was removed from the reaction mixture by centrifuge (13,000× g, 10 min). The supernatant was subjected to 19F-NMR analysis.
3. Results and Discussion
Considering that MA37 produces a broader spectrum of fluorometabolites compared to
S. cattleya, the gene homologues of
fthB and
fthC in MA37 (
fthBMA and
fthCMA, respectively) may have roles in other unidentified fluorometabolites. To this end, we generated two MA37 variants where
fthBMA and
fthCMA were subjected to in-frame deletion, respectively. After fermentation (50 mL, 28 °C, 12 d), the supernatants of the culture broths of the two variants along with the wild type (WT) were subjected to
19F-NMR analysis. The MA37_Δ
fthBMA variant demonstrated a relatively similar pattern of secreted
2 as well as other unidentified fluorometabolite production to the WT, consistent with the previous conclusion for
S.
cattleya [
9] (
Figure 2A,B).
Gene inactivation of
fthCMA, which encodes a putative 4-FT exporter, led to significant reduction of
2 as observed in
19F-NMR analysis, also consistent with the previous report [
9]. We noticed that
1 together with five other unknown fluorometabolites (compounds with star labels and 5-FDR
8,
9 (
Figure 2D)) showed increased production in the MA37_Δ
fthCMA variant compared to the ones in the WT (
Figure 2A,C and
Figure S1). The accumulation of these unidentified fluorometabolites in the MA37_Δ
fthCMA variant suggested that they are shunt metabolites, arising from aberrant derailment of the main pathways of
1 and
2 in MA37.
Next, we investigated the effect of the biosynthetic genes in the
fdr BGC on other fluorometabolites, which contain three functionally assigned proteins in the pathway of
3 [
2]. FdrA was proposed to be a metal-dependent phosphoesterase that mediates the dephosphorylation of 5-FDRP
11 to generate
8, the key intermediate in the second biosynthetic pathway leading to the production of
3 [
2]. FdrC has been biochemically characterized to be a short-chain dehydrogenase that catalyzes the oxidation of 5-FDR
8, followed by spontaneous hydrolysis to yield
3. FdrB is a putative dihydroxyacid dehydratase, an enzyme analogue of SalH in the biosynthesis of salinosporamide A [
19], which has been proposed to convert 5-chlororibonate to 5-chloro-4-hydroxy-2-oxopentanoate (
Figure S2).
To this end, we generated two MA37 variants, Δ
fdrA and Δ
fdrB. Comparative fluorometabolite profiling observed in the
19F-NMR analysis revealed that the production of
3 in these two variants was reduced compared to in the WT (
Figure 3) while
1 and
2 were only mildly affected. Interestingly, the inactivation of individual genes indeed affected other unidentified fluorometabolites. One of the fluorine signals with a significantly different intensity is found at −227.80 ppm (compound
13 in
Figure 3C,D). The production of this metabolite was abolished in Δ
fdrA and Δ
fdrB variants compared with the one in the WT, suggesting that the production of this metabolite is directly related to the catalytic functions of FdrA and FdrB. One possibility is that this metabolite is 5-fluoro-4-hydroxy-2-oxopentanoate
13, a dehydrated product generated by the action of FdrB on
3.
Another significant fluctuation in the
19F-NMR spectra is the signal at −231.42 ppm (compound
9 in
Figure 3). While the production level of this metabolite appears to be reduced in the Δ
fdrB variant compared to the WT (
Figure 3A,B), the inactivation of
fdrA upregulated the production of this metabolite as observed in
Figure 3C. Interestingly, this fluorometabolite was also accumulated in the Δ
fthCMA MA37 variant, suggesting that the synthesis of this fluorometabolite may result from the common intermediates in the biosynthetic pathways of
1,
2 and
3. However, the biochemical characterization of the enzymes produced by the genes
fdrA and
fdrB was not successful due to production problems in
E. coli (inclusion bodies). Based on previous studies in
S. cattleya, the chemical shift of this metabolite in
19F-NMR spectrum highly coincided with that of 5-FDRul
9 [
17]. Previously,
9 was synthesized to probe the fluorometabolism in
S. cattleya, and it was found that it is not one of the intermediates or a metabolite in the culture of
S. cattleya. To confirm whether
9 was indeed a metabolite in MA37, the compound was prepared synthetically according to the protocols in the literature [
17]. Moreover,
8 was first chemically prepared from D-ribose according to the literature (
Figure 4A and
Figures S3–S7) [
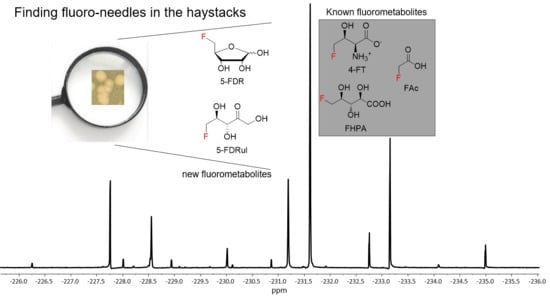
18], followed by enzymatic conversion to 5-FDRul using commercially available glucose isomerase (Sigma Aldrich UK cat no. G4166). The inspection of
19F-NMR indicated that
9 (−231.42 ppm observed in
19F-NMR) (
Figure 4C and
Figures S8 and S9) was indeed generated in a mixture of unreacted α- and β- anomers of
8 (−227.80 and −231.42 ppm, respectively) compared with the synthetic 5-FDR sample (
Figure 4B). The
19F-NMR signals coincided when the enzyme reaction sample was mixed with the supernatant of MA37 fermentation broth (
Figure 4E), confirming that the fluorine signal at −231.42 ppm is
9. Interestingly, the chemical shifts of both anomers of
8 in the reaction mixture also overlapped with two previously unknown signals in the supernatant of MA37 culture broth as evidenced in the
19F-NMR spectra (
Figure 4B,E). Taken together, this study identified
9 as one of the unidentified fluorometabolites in MA37, further extending the very small collection of this rare class of bacterial natural products.
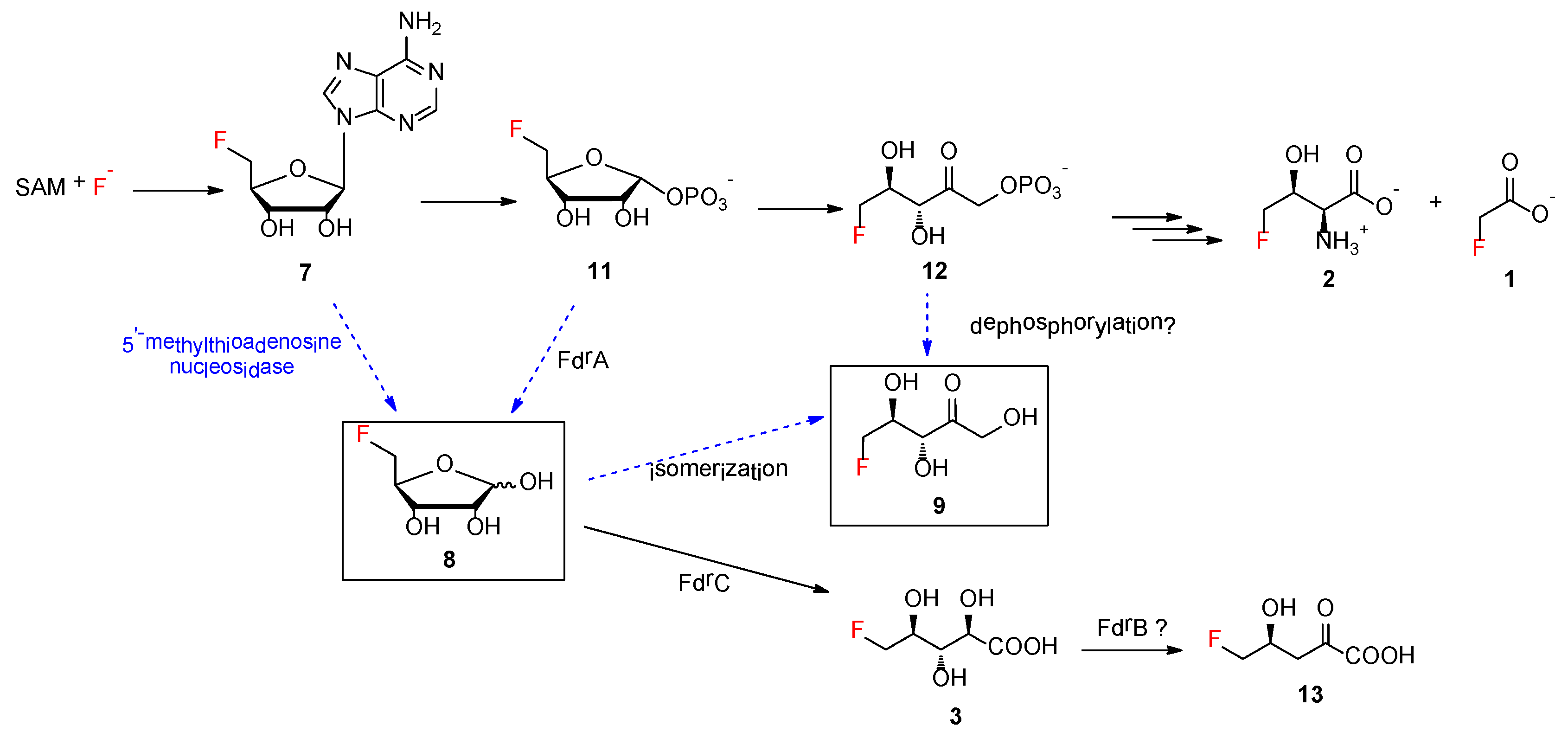
Additionally,
8 was proposed to be a key intermediate in the branched fluorometabolism for generating FHPA as a result of the action of FdrA, a metal-dependent phosphoesterase [
2]. The increased accumulation of
8 in the MA37 variants Δ
fdrA and Δ
fthCMA suggested that another promiscuous house-keeping enzyme in MA37 is able to generate
8. One possible enzyme candidate is 5′-methylthioadenosine (MTA) nucleosidase, a key enzyme, in the methionine salvage pathway in microbes, that catalyzes 5′-methylthioadenosine to its corresponding methylthioribose (
Figure 5) [
20]. Interestingly, it is likely that
S. cattleya may recruit 5-methylthio-ribose-1-phosphate isomerase, another key enzyme in the methionine salvage pathway, as part of its fluorometabolism toward
1 and
2 (
Figure S10) [
8]. This indicated that natural product biosynthetic pathways can recruit promiscuous enzymes from primary metabolisms to enable the evolution of biosynthetic capacity and expand the range of known organofluorine biochemistry. A BLAST search using the sequence of MTA nucleosidase from
Streptomyces laurentii (accession no. BAU88196) as a query against the annotated MA37 draft genome in the RAST server [
21] revealed the presence of one gene encoding MTA nucleosidase in MA37 (accession no. MT478135). The encoded protein possesses several crucial amino acids for substrate binding and catalytic function as displayed by multiple sequence alignment. Protein modelling in the Pyre2 server [
22] also indicated that the overall predicted structure shares high homologue (100% confidence) with other MTA nucleosidases and purine nucleoside hydrolases (
Figure S11). This analysis is in sharp contrast to what has been observed in
S. cattleya and
S. xinghaiensis. No such gene encoding MTA nucleosidase can be found in the genomes of either
S. cattleya or
S. xinghaiensis. It is known that
S. cattleya is the producer of
1 and
2 [
23] and that
S. xinghaiensis only produces
1 [
23]. In both cases, no other fluorometabolites were found in these bacteria. Our bioinformatics analysis strongly suggests that the putative MTA nucleosidase in MA37 is responsible for the increased accumulation of
8. MTA nucleosidases catalyze a similar biochemical reaction to that of the purine nucleoside hydrolase identified in the purine salvage pathway that catalyzes adenosine to D-ribose. Previously, a purine nucleoside hydrolase from
Trypanosoma vivax (TvNH) was shown to mediate the conversion of 5′-FDA to 5-FDR. As a result, it has been utilized as a biocatalyst in one-pot biotransformation involving fluorinase for the synthesis of 5-deoxy-5-[
18F] fluororibose as a potential diagnostic reagent for positron emission tomography applications [
24]. MTA nucleosidase from
E.
coli has been shown to be able to convert adenosine to D-ribose, albeit less efficiently than its natural substrate, 5′-methylthioadenosine [
25]. Therefore, the putative MTA nucleosidase in MA37 is likely to be responsible for the accumulation of
8 in the MA37 variants. The biochemical characterization of this protein may offer an alternative biocatalyst to the biotransformation of 5-deoxy-5-[
18F] fluororibose.
The bio-origin of
9 still remains to be biochemically determined. It is worth noting that, although
9 has been synthesized as a chemical probe,
9 is not part of the fluorometabolism in
S. cattleya [
17]. In the case of MA37,
9 is likely to be a shunt product deviating from the two main fluorometabolisms, resulting from the premature isomerization or hydrolysis of reactive intermediates, such as
8 or
12, respectively (
Figure 5). No gene encoding glucose isomerase was found in the draft genome of MA37. Although ribose isomerase, which has been shown to catalyze the conversion of D-ribose to D-ribulose, was purified from extracts of
mycobacterium smegatis in 1975 [
26], its protein sequence remains to be confirmed. One may not exclude the possibility that a homologue of ribose isomerase present in MA37 is responsible for the production of compound
9 in MA37 WT and its variants. Another possibility is that the presence of
9 may result from a promiscuous dephosphorylase enzyme that directly converts
12 to
9.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}