On the Risks of Phylogeny-Based Strain Prioritization for Drug Discovery: Streptomyces lunaelactis as a Case Study

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Ferroverdin A Quantification by High Pressure Liquid Chromatography (HPLC)
2.3. Compound Identification by Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry (UPLC–MS/MS)
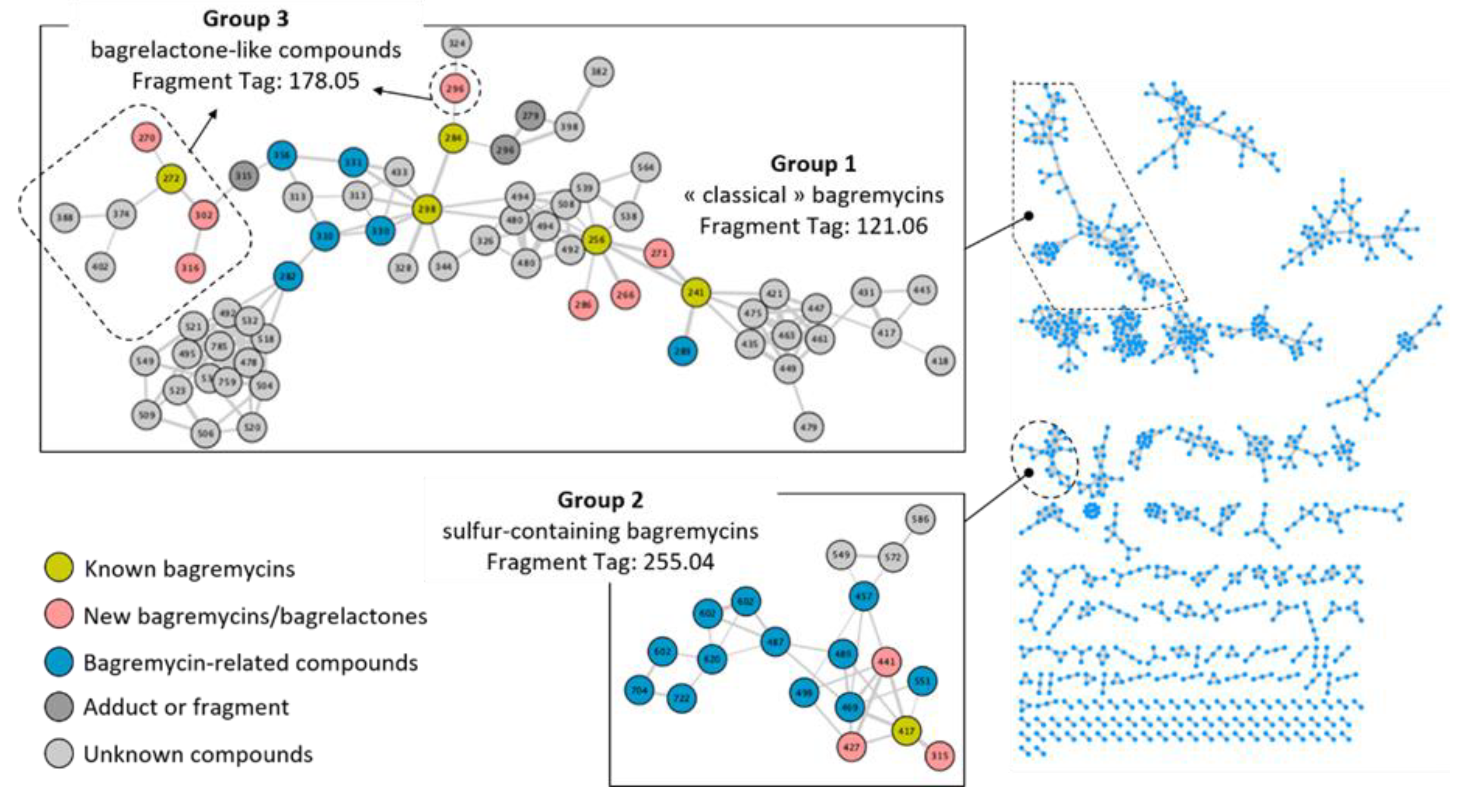
2.4. Molecular Network Construction
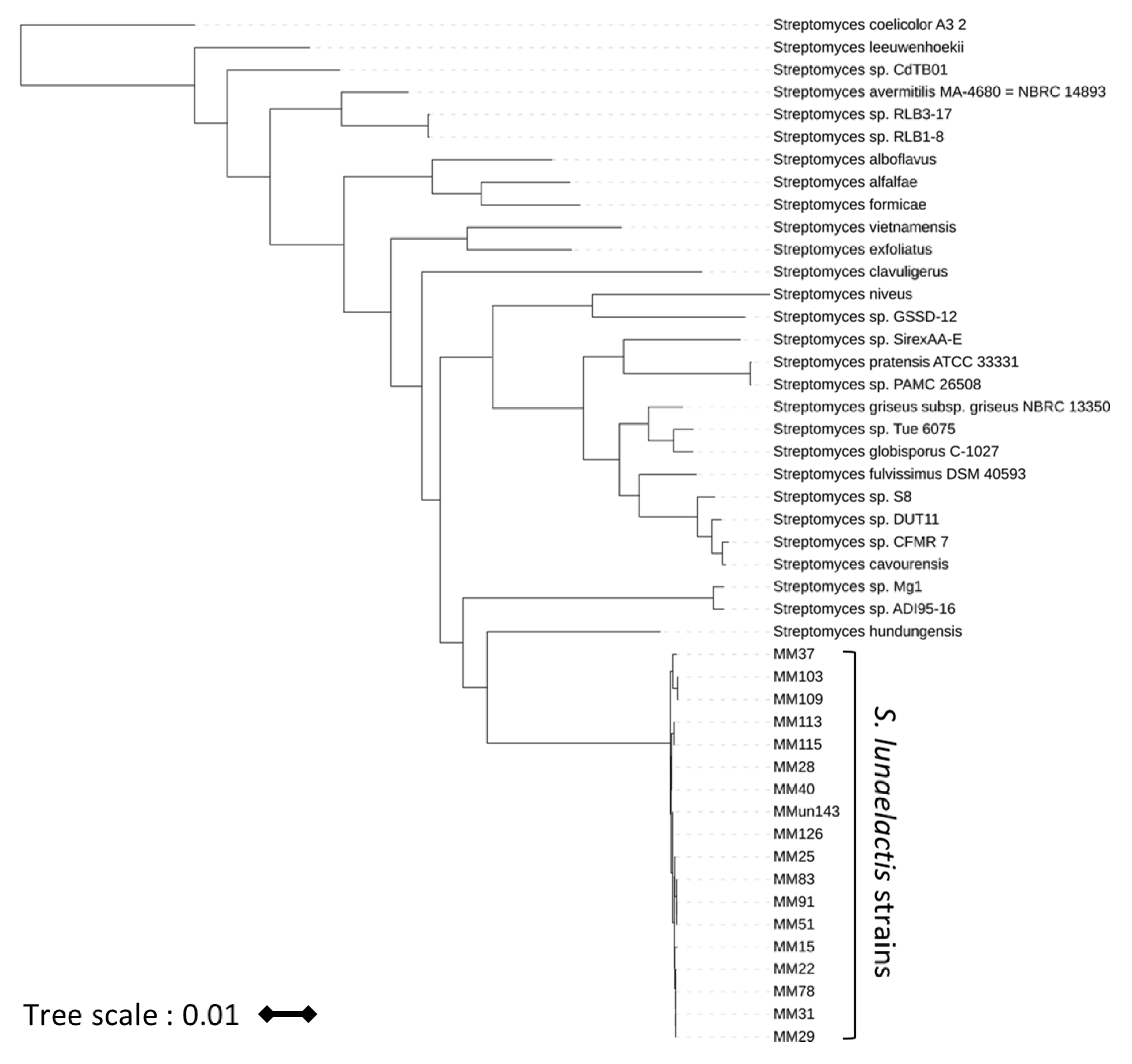
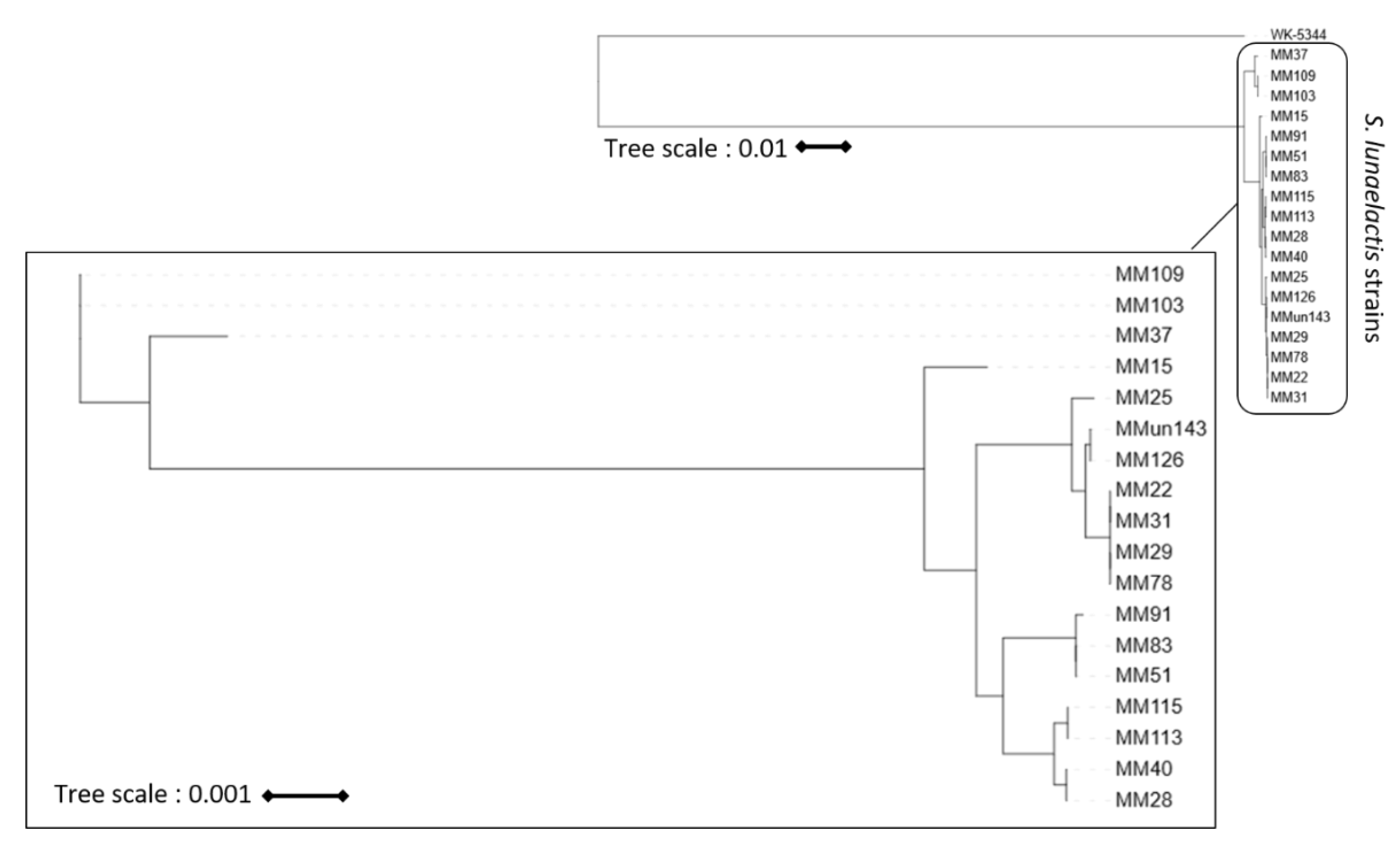
2.5. Phylogeny Analyses
2.6. Genome Sequencing
3. Results
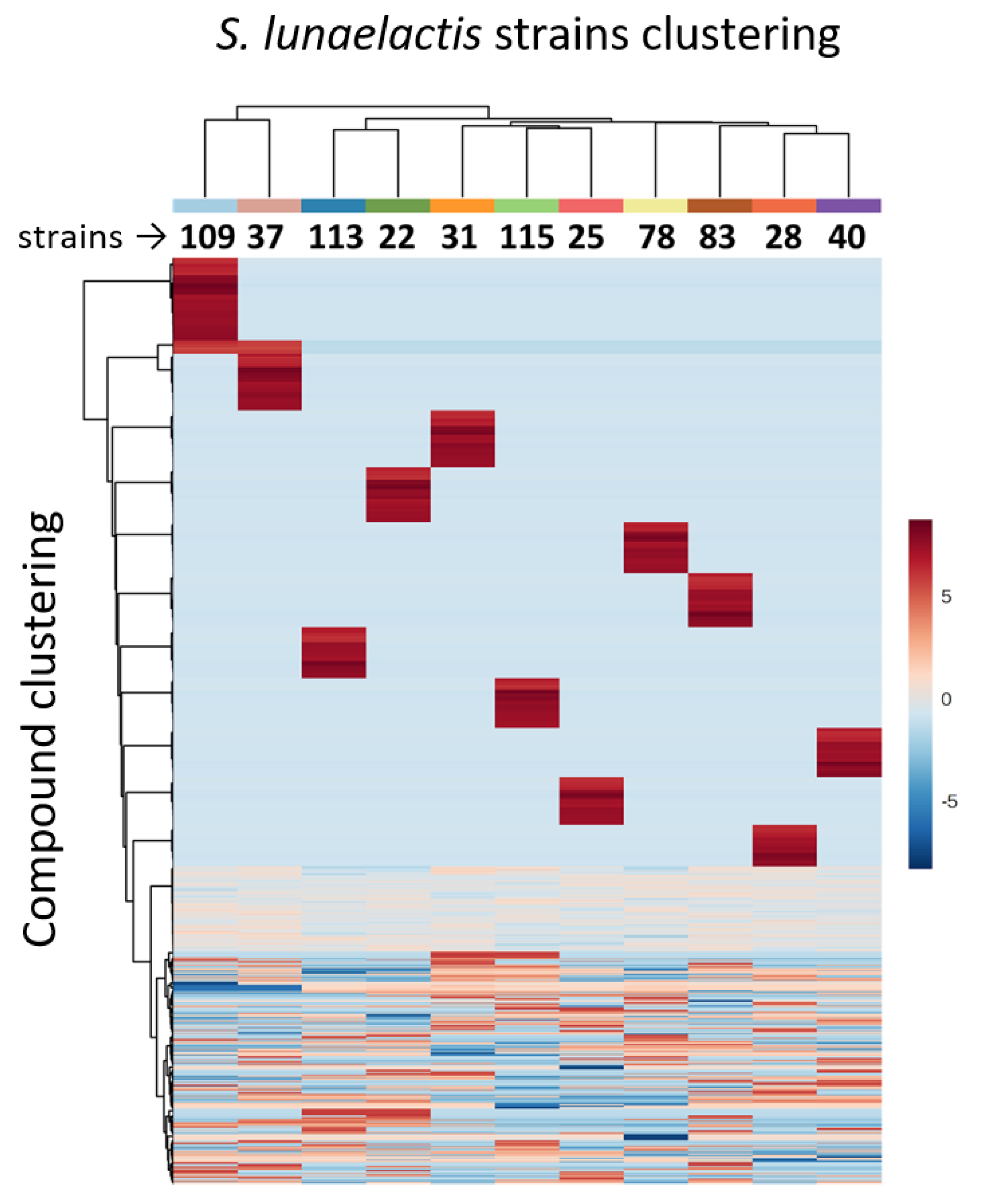
3.1. Genome Mining of 18 S. lunaelactis Strains Revealed Strain-Specific Secondary Metabolism
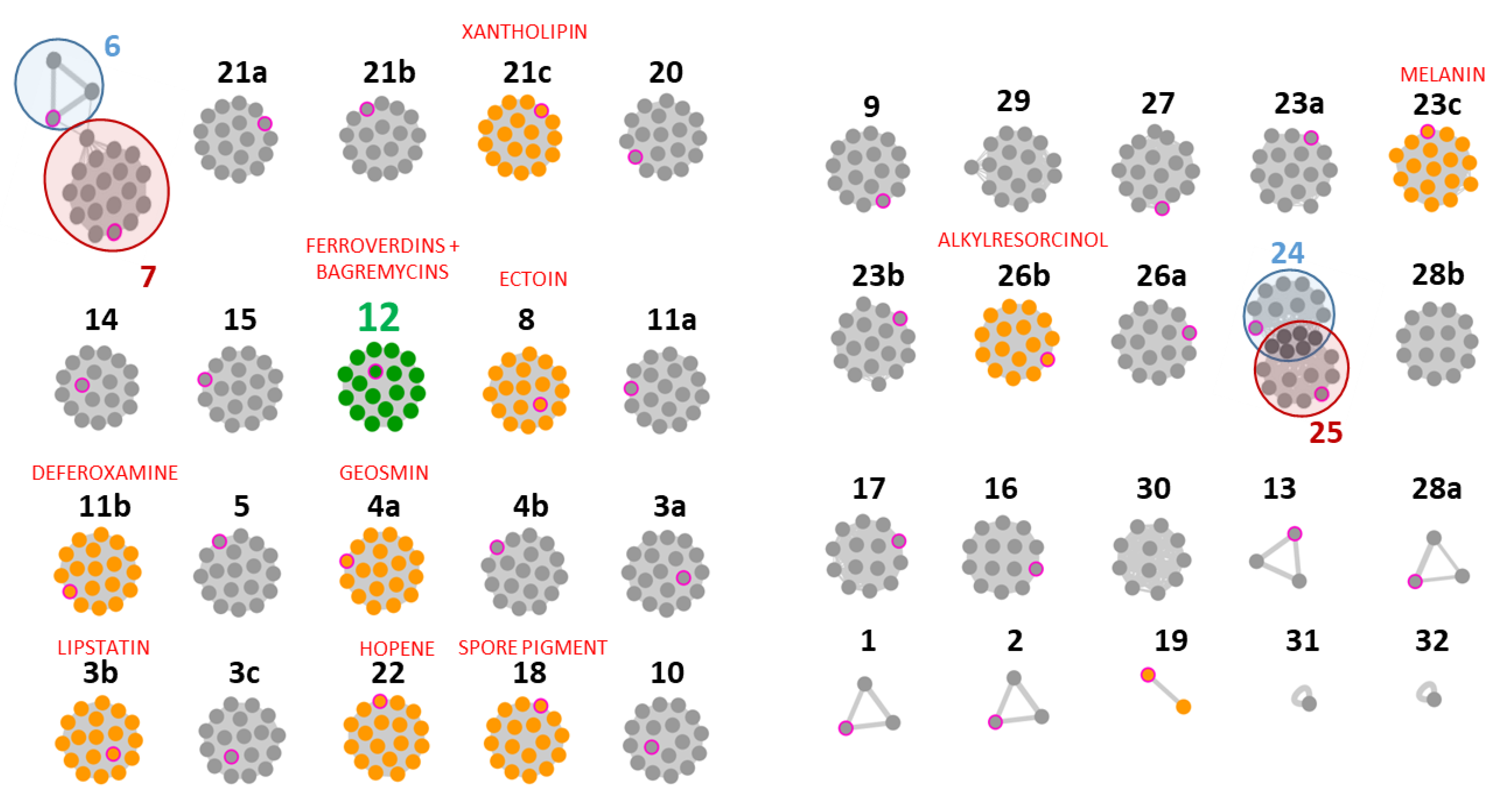
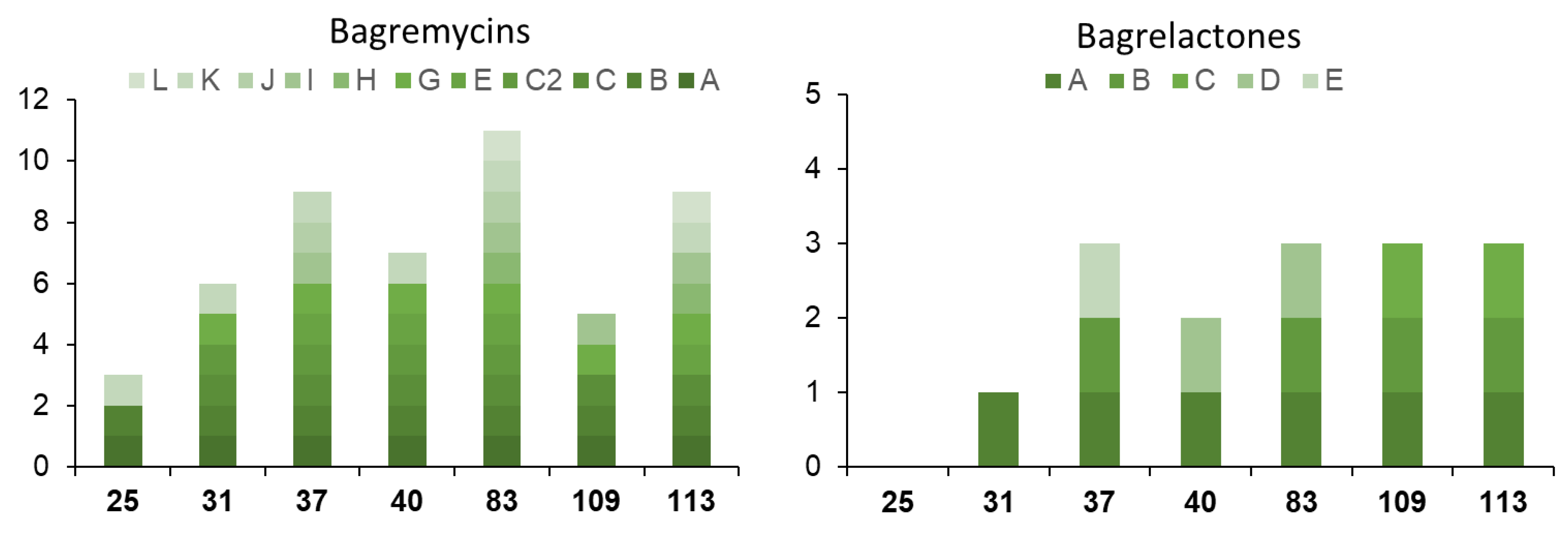
3.2. Strain-Specific Compound Diversity and Uneven Levels of Bagremycin and Ferroverdin Production Amongst the Various S. lunaelactis Strains
3.3. Heterogeneous Production of Bagremycins by S. lunaelactis Strains
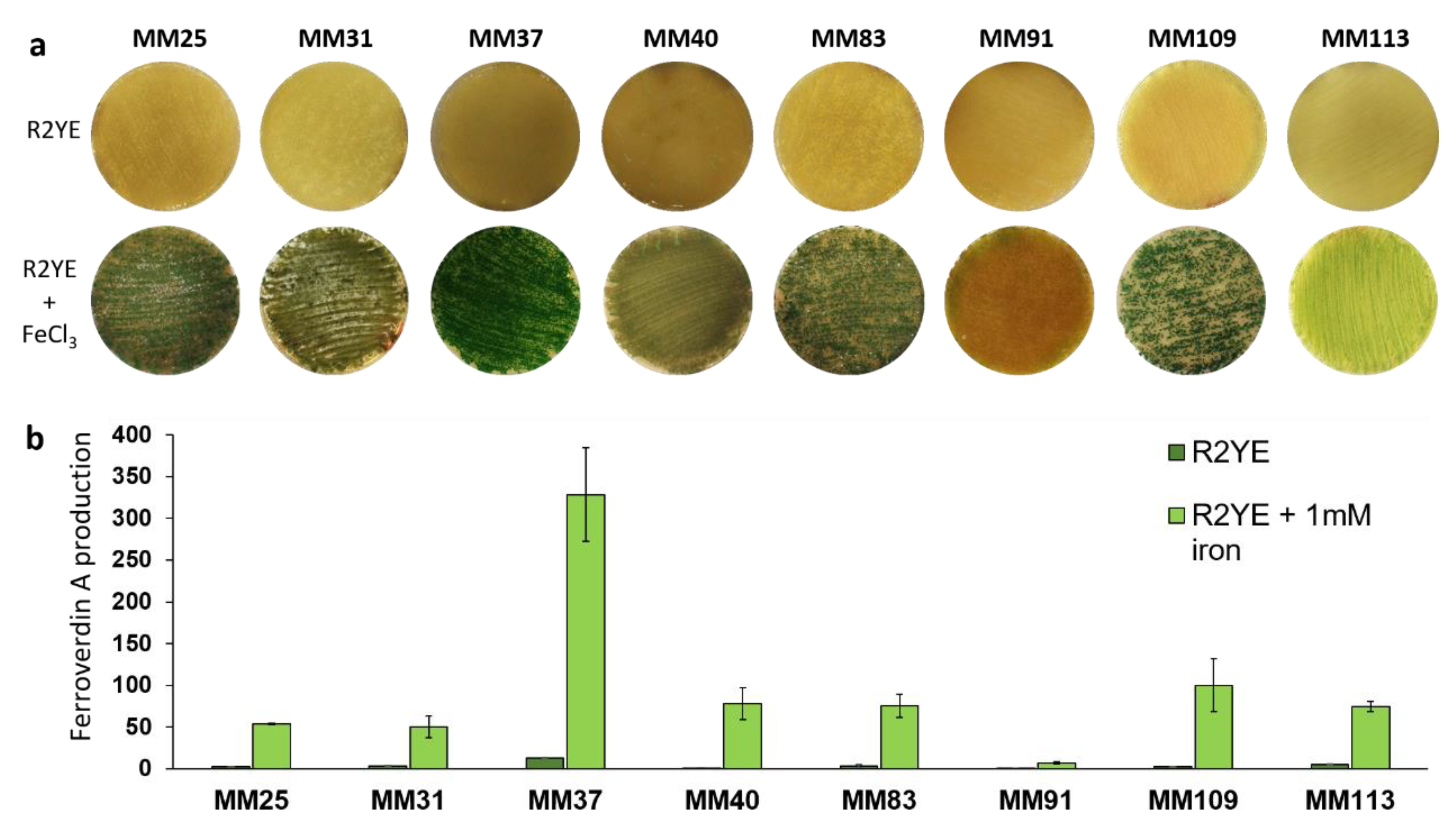
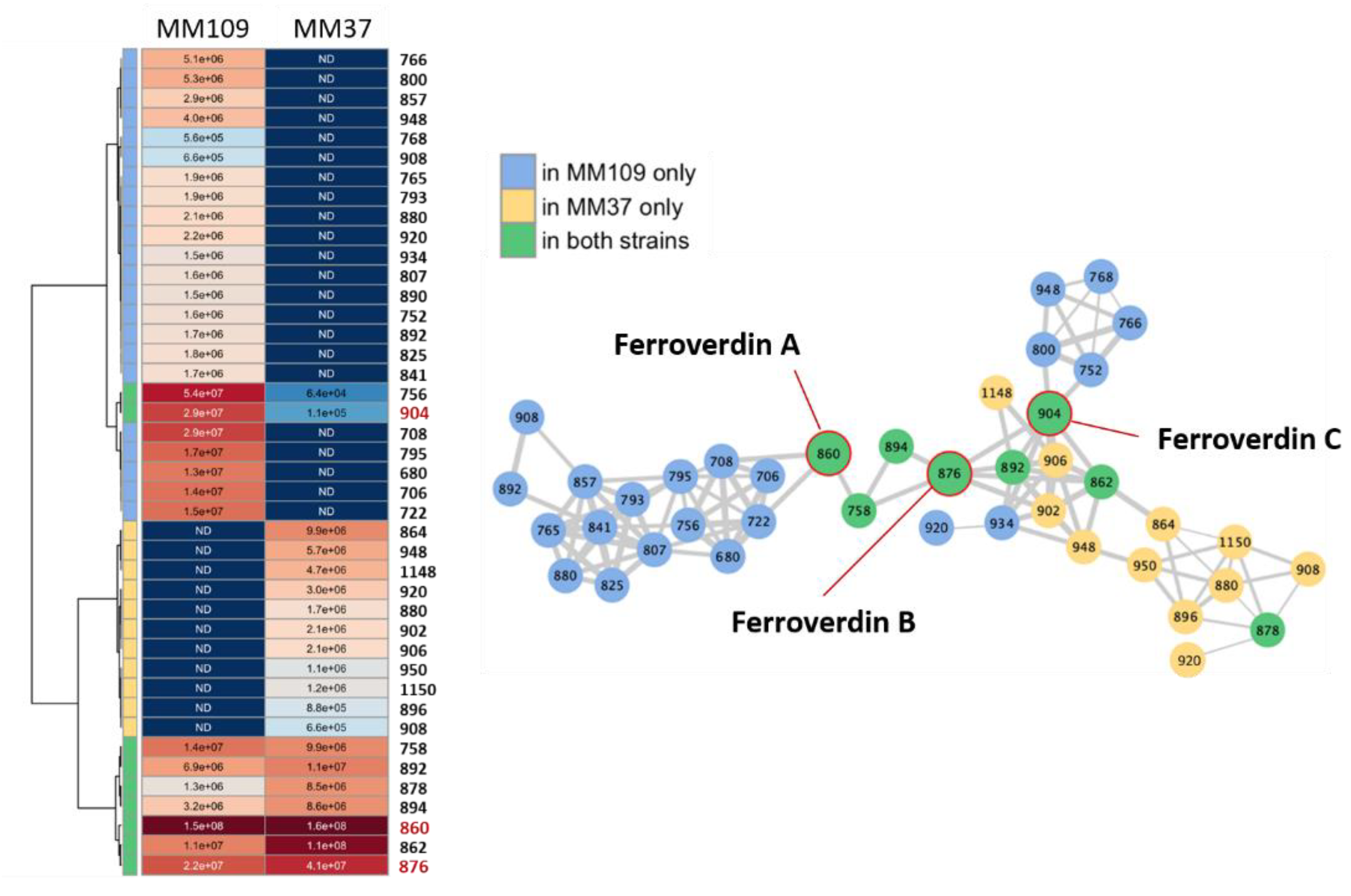
3.4. Heterogeneous Production Levels of Ferroverdins by S. lunaelactis Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Bull, A.T.; Goodfellow, M. Dark, rare and inspirational microbial matter in the extremobiosphere: 16 000 m of bioprospecting campaigns. Microbiology 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodfellow, M.; Nouioui, I.; Sanderson, R.; Xie, F.; Bull, A. Rare Taxa and Dark Microbial Matter: Novel Bioactive Actinobacteria Abound in Atacama Desert Soils. Available online: https://pubmed.ncbi.nlm.nih.gov/29721711/ (accessed on 21 May 2020).
- Hou, Y.; Braun, D.R.; Michel, C.R.; Klassen, J.L.; Adnani, N.; Wyche, T.P.; Bugni, T.S. Microbial strain prioritization using metabolomics tools for the discovery of natural products. Anal. Chem. 2012, 84, 4277–4283. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication strategies in natural product research: How many tools and methodologies behind the same concept? Phytochem. Rev. 2015, 16. [Google Scholar] [CrossRef]
- Demirev, P.; Sandrin, T.R. Applications of Mass Spectrometry in Microbiology: From Strain Characterization to Rapid Screening for Antibiotic Resistance; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-26070-9. [Google Scholar]
- Gaudêncio, S.P.; Pereira, F. Dereplication: Racing to speed up the natural products discovery process. Nat. Prod. Rep. 2015, 32, 779–810. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Ando, K.; Takagi, M.; Shin-ya, K. Dereplication of Streptomyces Strains by Automated Southern Hybridization with a Polyketide Synthase Gene Probe. Actinomycetologica 2010, 24, 66–69. [Google Scholar] [CrossRef]
- Park, C.J.; Andam, C.P. Within-Species Genomic Variation and Variable Patterns of Recombination in the Tetracycline Producer Streptomyces rimosus. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Seipke, R.F. Strain-Level Diversity of Secondary Metabolism in Streptomyces albus. PLoS ONE 2015, 10, e0116457. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.M.; Thibessard, A.; Lorenzi, J.-N.; Benhadj, M.; Hôtel, L.; Gacemi-Kirane, D.; Lespinet, O.; Leblond, P.; Aigle, B. Comparative Genomics among Closely Related Streptomyces Strains Revealed Specialized Metabolite Biosynthetic Gene Cluster Diversity. Antibiotics 2018, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Labeda, D.P.; Goodfellow, M.; Brown, R.; Ward, A.C.; Lanoot, B.; Vanncanneyt, M.; Swings, J.; Kim, S.-B.; Liu, Z.; Chun, J.; et al. Phylogenetic study of the species within the family Streptomycetaceae. Antonie Van Leeuwenhoek 2012, 101, 73–104. [Google Scholar] [CrossRef]
- Nouioui, I.; Carro, L.; García-López, M.; Meier-Kolthoff, J.P.; Woyke, T.; Kyrpides, N.C.; Pukall, R.; Klenk, H.-P.; Goodfellow, M.; Göker, M. Genome-Based Taxonomic Classification of the Phylum Actinobacteria. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Tidjani, A.-R.; Lorenzi, J.-N.; Toussaint, M.; van Dijk, E.; Naquin, D.; Lespinet, O.; Bontemps, C.; Leblond, P. Massive Gene Flux Drives Genome Diversity between Sympatric Streptomyces Conspecifics. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, D.; Maciejewska, M.; Naômé, A.; Martinet, L.; Coppieters, W.; Karim, L.; Baurain, D.; Rigali, S. Isolation, Characterization, and Antibacterial Activity of Hard-to-Culture Actinobacteria from Cave Moonmilk Deposits. Antibiotics 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewska, M.; Adam, D.; Martinet, L.; Naômé, A.; Całusińska, M.; Delfosse, P.; Carnol, M.; Barton, H.A.; Hayette, M.-P.; Smargiasso, N.; et al. A Phenotypic and Genotypic Analysis of the Antimicrobial Potential of Cultivable Streptomyces Isolated from Cave Moonmilk Deposits. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewska, M.; Pessi, I.S.; Arguelles-Arias, A.; Noirfalise, P.; Luis, G.; Ongena, M.; Barton, H.; Carnol, M.; Rigali, S. Streptomyces lunaelactis sp. nov., a novel ferroverdin A-producing Streptomyces species isolated from a moonmilk speleothem. Antonie Van Leeuwenhoek 2015, 107, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.; Lambert, S.; Jourdan, S.; Tenconi, E.; Colson, S.; Maciejewska, M.; Ongena, M.; Martin, J.F.; van Wezel, G.; Rigali, S. Unsuspected control of siderophore production by N-acetylglucosamine in streptomycetes. Environ. Microbiol. Rep. 2012, 4, 512–521. [Google Scholar] [CrossRef]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Martinet, L.; Naômé, A.; Deflandre, B.; Maciejewska, M.; Tellatin, D.; Tenconi, E.; Smargiasso, N.; de Pauw, E.; van Wezel, G.P.; Rigali, S. A Single Biosynthetic Gene Cluster Is Responsible for the Production of Bagremycin Antibiotics and Ferroverdin Iron Chelators. mBio 2019, 10, e01230-19. [Google Scholar] [CrossRef] [Green Version]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A Cross-platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with GNPS. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. BIOLOGICAL NETWORK EXPLORATION WITH CYTOSCAPE 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Naômé, A.; Maciejewska, M.; Calusinska, M.; Martinet, L.; Anderssen, S.; Adam, D.; Tenconi, E.; Deflandre, B.; Coppieters, W.; Karim, L.; et al. Complete Genome Sequence of Streptomyces lunaelactis MM109T, Isolated from Cave Moonmilk Deposits. Genome Announc. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Bertasso, M.; Holzenkämpfer, M.; Zeeck, A.; Dall’Antonia, F.; Fiedler, H.P. Bagremycin A and B, novel antibiotics from Streptomyces sp. Tü 4128. J. Antibiot. 2001, 54, 730–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chai, W.; Wang, W.; Song, T.; Lian, X.-Y.; Zhang, Z. Cytotoxic Bagremycins from Mangrove-Derived Streptomyces sp. Q22. J. Nat. Prod. 2017, 80, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Shu, C.; Lian, X.; Zhang, Z. New Antibacterial Bagremycins F and G from the Marine-Derived Streptomyces sp. ZZ745. Mar. Drugs 2018, 16, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, G.E. The Paradox of the Plankton. Am. Nat. 1961, 95, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Maciejewska, M.; Adam, D.; Naômé, A.; Martinet, L.; Tenconi, E.; Całusińska, M.; Delfosse, P.; Hanikenne, M.; Baurain, D.; Compère, P.; et al. Assessment of the Potential Role of Streptomyces in Cave Moonmilk Formation. Front. Microbiol. 2017, 8, 1181. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Name | Molecular Formula | m/z (exp) | Δm (ppm) | Producing Strains | Group | Refer. |
|---|---|---|---|---|---|---|---|
| 1 | Bagremycin A | C15H14NO3+ | 256.0967 | 0.4 | 113,25,31,83,40, 37,109 | 1 | [34] |
| 2 | Bagremycin B | C17H14NO4+ | 298.1074 | 0.1 | 113,25,31,83,40, 37,109 | 1 | [34] |
| 3 | Bagremycin C | C20H20N2O6S+ | 417.1115 | 0.1 | 113,31,83,40,37, 109 | 2 | [35] |
| 4 | Bagremycin E | C15H12O3+ | 241.0859 | 0.3 | 113,83,40,37 | 1 | [35] |
| 5 | Bagremycin G | C16H13NO4+ | 284.0915 | 0.8 | 113,31,83,40,37, 109 | 1 | [35] |
| 6 | Bagremycin H | C16H14O4+ | 271.0964 | 0.2 | 113,83 | 1 (NL) | This work |
| 7 | Bagremycin I | C16H11NO3+ | 266.0812 | 0.1 | 113,83,37,109 | 1 (NL) | This study |
| 8 | Bagremycin J | C15H12NO3S+ | 286.0531 | 0.4 | 83,37 | 1 (NL) | This study |
| 9 | Bagremycin C2 | C11H8N2O6S+ | 315.0643 | 0.7 | 31,83,40,37 | 2 | This study |
| 10 | Bagremycin K | C21H18N2O6S+ | 427.0959 | 0.3 | 113,25,31,83,40,37 | ND | This study |
| 11 | Bagremycin L | C22H20N2O6S+ | 441.1118 | 0.5 | 113,83 | ND | This study |
| 12 | Bagrelactone A | C15H13NO4+ | 272.0922 | 0.2 | 113,31,83,40,37, 109 | 3 | [35] |
| 13 | Bagrelactone B | C17H13NO4+ | 296.0916 | 0.2 | 83,37,109,113 | ND * | This study |
| 14 | Bagrelactone C | C16H15NO3+ | 270.1125 | 0.1 | 113,109 | ND * | This study |
| 15 | Bagrelactone D | C16H15NO5+ | 302.1023 | 0.1 | 83,40 | 3 | This study |
| 16 | Bagrelactone E | C17H17NO5+ | 316.1179 | 0.3 | 37 | 3 | This study |
| From compounds 17 to 33, MS/MS fragmentation did not allow us to predict the structure. | |||||||
| 17 | C19H18NO6+ | 356.1127 | 0.5 | 37,109 | 3 | This study | |
| 18 | C15H20NO6+ | 310.1285 | 0.1 | 25,83,37,109 | 3 | This study | |
| 19 | C16H15N2O6+ | 331.0919 | 0.8 | 113,25,40 | 3 | This study | |
| 20 | C17H16NO6+ | 330.0969 | 0.8 | 25,83,40,37,109 | 3 | This study | |
| 21 | C13H16NO6+ | 282.0972 | 0.1 | 113,25,31,83,37, 109 | 3 | This study | |
| 22 | C16H21 N2O3+ | 289.1546 | 0.3 | 109 | 1 (NL) | This study | |
| 23 | C28H27N2O8S + | 551.1484 | 0.3 | 83 | 2 | This study | |
| 24 | C23H21N2O7S + | 469.1066 | 0.4 | 113,83 | ND | This study | |
| 25 | C24H23N2O8S + | 499.1170 | 0.1 | 113,25,31,83,109 | ND | This study | |
| 26 | C23H25N2O8S + | 489.1328 | 0.3 | 113,31,83,40,109 | 2 | This study | |
| 27 | C22H21N2O7S+ | 457.1063 | 0.2 | 113,31,83,40,37, 109 | ND | This study | |
| 28 | C23H23N2O8S + | 487.1171 | 0.3 | 113,83 | ND | This study | |
| 29 | C31H28N3O8S + | 602.1587 | 0.7 | 25,31,37,109, 113,83 | 2 | This study | |
| 30 | C30H23N3O9S + | 602.1231 | 0.6 | 113,83 | ND | This study | |
| 31 | C30H25N3O10S + | 620.1336 | 0.1 | 31,37,40,83,109, 113 | 2 | This study | |
| 32 | C38H32N3O10S + | 722.1799 | 0.5 | 113,83 | 2 | This study | |
| 33 | C38H30N3O9S + | 704.1696 | 0.2 | 113,83 | 2 | This study | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinet, L.; Naômé, A.; Baiwir, D.; De Pauw, E.; Mazzucchelli, G.; Rigali, S. On the Risks of Phylogeny-Based Strain Prioritization for Drug Discovery: Streptomyces lunaelactis as a Case Study. Biomolecules 2020, 10, 1027. https://doi.org/10.3390/biom10071027
Martinet L, Naômé A, Baiwir D, De Pauw E, Mazzucchelli G, Rigali S. On the Risks of Phylogeny-Based Strain Prioritization for Drug Discovery: Streptomyces lunaelactis as a Case Study. Biomolecules. 2020; 10(7):1027. https://doi.org/10.3390/biom10071027
Chicago/Turabian StyleMartinet, Loïc, Aymeric Naômé, Dominique Baiwir, Edwin De Pauw, Gabriel Mazzucchelli, and Sébastien Rigali. 2020. "On the Risks of Phylogeny-Based Strain Prioritization for Drug Discovery: Streptomyces lunaelactis as a Case Study" Biomolecules 10, no. 7: 1027. https://doi.org/10.3390/biom10071027