Reduced Reelin Expression in the Hippocampus after Traumatic Brain Injury


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Handling
2.2. Controlled Cortical Impact (CCI) Injury Model
2.3. Brain Tissue Section Preparation
2.4. Immunofluorescence Assays
2.5. mRNA Isolation and Quantitative Reverse Transcription PCR (RT-qPCR) Analysis
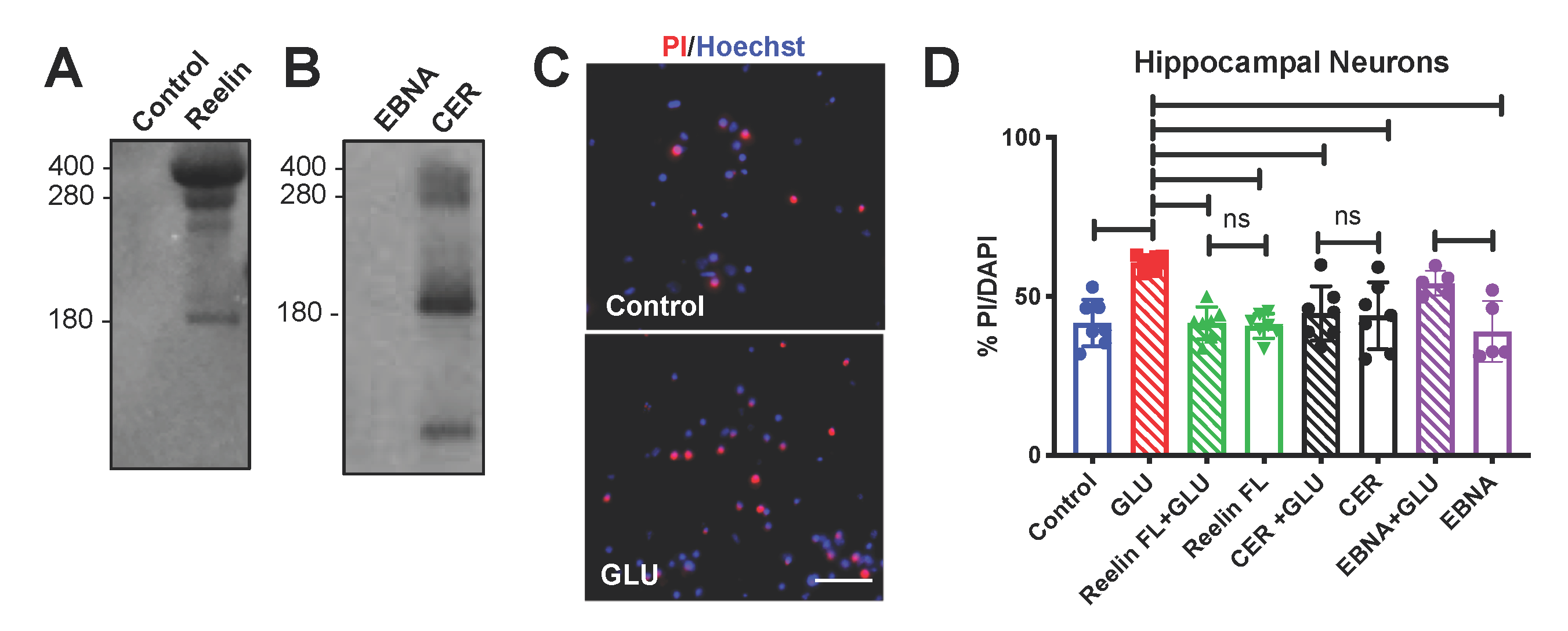
2.6. Reelin Protein Expression and Purification
2.7. Dissociated Hippocampal Neurons
2.8. Cell Death Staining and Analysis
2.9. Statistical Analysis
3. Results
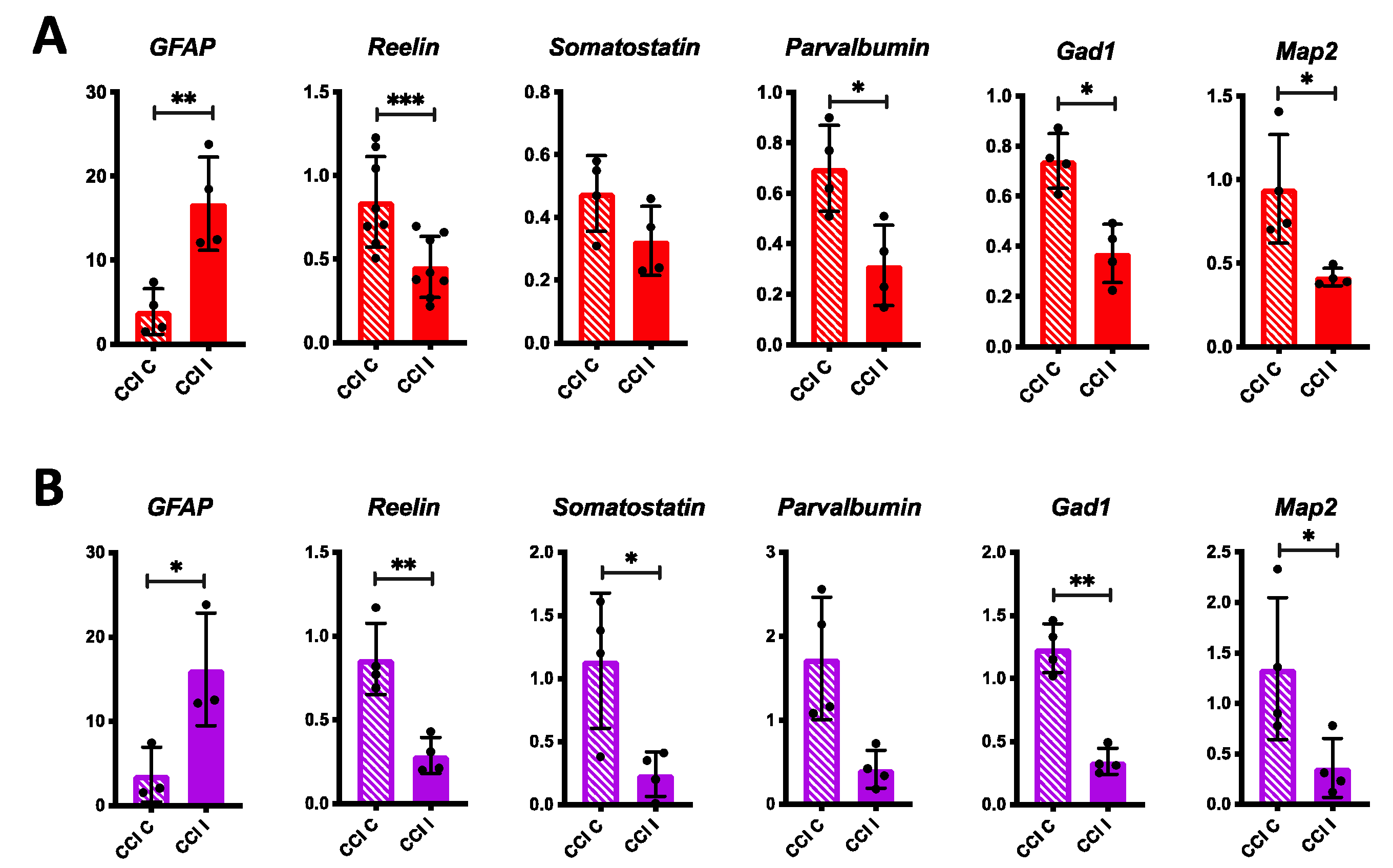
3.1. Short-Term Effects of CCI on Inflammation, Reelin, and Neuronal Markers Expression in the Mouse Forebrain
3.2. Medium and Long-Term Effects of CCI on Reelin Expression in the Mouse Forebrain
3.3. Neuroprotective Effect of Reelin in Hippocampal Neurons
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Humphreys, I.; Wood, R.L.; Phillips, C.J.; Macey, S. The costs of traumatic brain injury: A literature review. Clin. Outcomes Res. 2013, 5, 281–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyder, A.A.; Wunderlich, C.A.; Puvanachandra, P.; Gururaj, G.; Kobusingye, O.C. The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation 2007, 22, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Prim. 2016, 2, 16084. [Google Scholar] [CrossRef] [PubMed]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of traumatic brain injury: Time for a paradigm shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Mahmood, A.; Chopp, M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin. J. Traumatol. 2018, 21, 137–151. [Google Scholar] [CrossRef]
- Barrett, E.C.; McBurney, M.I.; Ciappio, E.D. Omega-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv. Nutr. 2014, 5, 268–277. [Google Scholar] [CrossRef]
- Mazzeo, A.T.; Beat, A.; Singh, A.; Bullock, M.R. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after tbi. Exp. Neurol. 2009, 218, 363–370. [Google Scholar] [CrossRef]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal models of traumatic brain injury and assessment of injury severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef]
- Romine, J.; Gao, X.; Chen, J. Controlled cortical impact model for traumatic brain injury. J. Vis. Exp. 2014, 90, e51781. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Mao, H. Quantifying the effect of repeated impacts and lateral tip movements on brain responses during controlled cortical impact. J. Neurotrauma 2019, 36, 1828–1835. [Google Scholar] [CrossRef]
- Osier, N.; Dixon, C.E. The controlled cortical impact model of experimental brain trauma: Overview, research applications, and protocol. Methods Mol. Biol. 2016, 1462, 177–192. [Google Scholar]
- Siebold, L.; Obenaus, A.; Goyal, R. Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp. Neurol. 2018, 310, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Sullivan, P.G.; Gibson, T.R.; Pavel, K.M.; Thompson, B.M.; Scheff, S.W. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: More than a focal brain injury. J. Neurotrauma 2005, 22, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Adelson, P.D.; Fellows-Mayle, W.; Kochanek, P.M.; Dixon, C.E. Morris water maze function and histologic characterization of two age-at-injury experimental models of controlled cortical impact in the immature rat. Childs Nerv. Syst. 2013, 29, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mao, H.; Yang, K.H.; Abel, T.; Meaney, D.F. A modified controlled cortical impact technique to model mild traumatic brain injury mechanics in mice. Front. Neurol. 2014, 5, 100. [Google Scholar] [CrossRef] [Green Version]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef]
- Lambert de Rouvroit, C.; Goffinet, A.M. The reeler mouse as a model of brain development. Adv. Anat Embryol. Cell. Biol. 1998, 150, 1–108. [Google Scholar]
- Wasser, C.R.; Herz, J. Reelin: Neurodevelopmental architect and homeostatic regulator of excitatory synapses. J. Biol. Chem. 2017, 292, 1330–1338. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; D’Arcangelo, G. New insights into reelin-mediated signaling pathways. Front. Cell. Neurosci. 2016, 10, 122. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid beta toxicity in vivo. Sci. Signal 2015, 8, ra67. [Google Scholar] [CrossRef] [Green Version]
- Israelsson, C.; Wang, Y.; Kylberg, A.; Pick, C.G.; Hoffer, B.J.; Ebendal, T. Closed head injury in a mouse model results in molecular changes indicating inflammatory responses. J. Neurotrauma 2009, 26, 1307–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.J.; Templet, S.; Zemoura, K.; Kuzniewska, B.; Pena, F.X.; Hwang, H.; Lei, D.J.; Haensgen, H.; Nguyen, S.; Saenz, C.; et al. Rapid, experience-dependent translation of neurogranin enables memory encoding. Proc. Natl. Acad. Sci. USA 2018, 115, E5805–E5814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, C.; Nitta, N.; Flubacher, A.; Muller, M.; Fahrner, A.; Kirsch, M.; Freiman, T.; Suzuki, F.; Depaulis, A.; Frotscher, M.; et al. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J. Neurosci. 2006, 26, 4701–4713. [Google Scholar] [CrossRef] [PubMed]
- Filice, F.; Vorckel, K.J.; Sungur, A.O.; Wohr, M.; Schwaller, B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing gaba interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 2016, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Cordoba-Chacon, J.; Gahete, M.D.; Castano, J.P.; Kineman, R.D.; Luque, R.M. Somatostatin and its receptors contribute in a tissue-specific manner to the sex-dependent metabolic (fed/fasting) control of growth hormone axis in mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E46–E54. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin promotes hippocampal dendrite development through the vldlr/apoer2-dab1 pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; Chhangawala, Z.; von Daake, S.; Savas, J.N.; Yates, J.R., 3rd; Comoletti, D.; D’Arcangelo, G. Reelin induces erk1/2 signaling in cortical neurons through a non-canonical pathway. J. Biol. Chem. 2014, 289, 20307–20317. [Google Scholar] [CrossRef] [Green Version]
- Israelsson, C.; Bengtsson, H.; Lobell, A.; Nilsson, L.N.; Kylberg, A.; Isaksson, M.; Wootz, H.; Lannfelt, L.; Kullander, K.; Hillered, L.; et al. Appearance of cxcl10-expressing cell clusters is common for traumatic brain injury and neurodegenerative disorders. Eur. J. Neurosci. 2010, 31, 852–863. [Google Scholar] [CrossRef]
- Okonkwo, D.O.; Yue, J.K.; Puccio, A.M.; Panczykowski, D.M.; Inoue, T.; McMahon, P.J.; Sorani, M.D.; Yuh, E.L.; Lingsma, H.F.; Maas, A.I.; et al. Gfap-bdp as an acute diagnostic marker in traumatic brain injury: Results from the prospective transforming research and clinical knowledge in traumatic brain injury study. J. Neurotrauma 2013, 30, 1490–1497. [Google Scholar] [CrossRef]
- Cantu, D.; Walker, K.; Andresen, L.; Taylor-Weiner, A.; Hampton, D.; Tesco, G.; Dulla, C.G. Traumatic brain injury increases cortical glutamate network activity by compromising gabaergic control. Cereb. Cortex 2015, 25, 2306–2320. [Google Scholar] [CrossRef] [Green Version]
- Alcantara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; de Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and cellular patterns of reelin mrna expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesold, C.; Impagnatiello, F.; Pisu, M.G.; Uzunov, D.P.; Costa, E.; Guidotti, A.; Caruncho, H.J. Reelin is preferentially expressed in neurons synthesizing g-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA 1998, 95, 3221–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jossin, Y.; Ignatova, N.; Hiesberger, T.; Herz, J.; Lambert de Rouvroit, C.; Goffinet, A.M. The central fragment of reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J. Neurosci. 2004, 24, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Yasui, N.; Kitago, Y.; Beppu, A.; Kohno, T.; Morishita, S.; Gomi, H.; Nagae, M.; Hattori, M.; Takagi, J. Functional importance of covalent homodimer of reelin protein linked via its central region. J. Biol. Chem. 2011, 286, 35247–35256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakage, Y.; Kato, M.; Hongo, A.; Ogino, H.; Ishii, K.; Ishizuka, T.; Kamei, T.; Tsuiji, H.; Miyamoto, T.; Oishi, H.; et al. A disintegrin and metalloproteinase with thrombospondin motifs 2 cleaves and inactivates reelin in the postnatal cerebral cortex and hippocampus, but not in the cerebellum. Mol. Cell. Neurosci. 2019, 100, 103401. [Google Scholar] [CrossRef]
- Courtes, S.; Vernerey, J.; Pujadas, L.; Magalon, K.; Cremer, H.; Soriano, E.; Durbec, P.; Cayre, M. Reelin controls progenitor cell migration in the healthy and pathological adult mouse brain. PLoS ONE 2011, 6, e20430. [Google Scholar] [CrossRef] [Green Version]
- Pulido, J.S.; Sugaya, I.; Comstock, J.; Sugaya, K. Reelin expression is upregulated following ocular tissue injury. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 889–893. [Google Scholar] [CrossRef]
- Turtzo, L.C.; Budde, M.D.; Gold, E.M.; Lewis, B.K.; Janes, L.; Yarnell, A.; Grunberg, N.E.; Watson, W.; Frank, J.A. The evolution of traumatic brain injury in a rat focal contusion model. NMR Biomed. 2013, 26, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Cullen, K.M.; Kocsi, Z.; Stone, J. Pericapillary haem-rich deposits: Evidence for microhaemorrhages in aging human cerebral cortex. J. Cereb. Blood Flow Metab. 2005, 25, 1656–1667. [Google Scholar] [CrossRef] [Green Version]
- Stary, C.M.; Xu, L.; Sun, X.; Ouyang, Y.B.; White, R.E.; Leong, J.; Li, J.; Xiong, X.; Giffard, R.G. Microrna-200c contributes to injury from transient focal cerebral ischemia by targeting reelin. Stroke 2015, 46, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Atif, H.; Hicks, S.D. A review of microrna biomarkers in traumatic brain injury. J. Exp. Neurosci. 2019, 13, 1179069519832286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, J.; Bjorklund, G.R.; Newbern, J.; Anderson, T. Parvalbumin fast-spiking interneurons are selectively altered by paediatric traumatic brain injury. J. Physiol. 2018, 596, 1277–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brizuela, M.; Blizzard, C.A.; Chuckowree, J.A.; Pitman, K.A.; Young, K.M.; Dickson, T. Mild traumatic brain injury leads to decreased inhibition and a differential response of calretinin positive interneurons in the injured cortex. J. Neurotrauma 2017, 34, 2504–2517. [Google Scholar] [CrossRef]
- Pesold, C.; Liu, W.S.; Guidotti, A.; Costa, E.; Caruncho, H.J. Cortical bitufted, horizontal, and martinotti cells preferentially express and secrete reelin into perineuronal nets, nonsynaptically modulating gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 3217–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates nmda receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef]
- Sinagra, M.; Verrier, D.; Frankova, D.; Korwek, K.M.; Blahos, J.; Weeber, E.J.; Manzoni, O.J.; Chavis, P. Reelin, very-low-density lipoprotein receptor, and apolipoprotein e receptor 2 control somatic nmda receptor composition during hippocampal maturation in vitro. J. Neurosci. 2005, 25, 6127–6136. [Google Scholar] [CrossRef] [Green Version]
- Ventruti, A.; Kazdoba, T.M.; Niu, S.; D’Arcangelo, G. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 2011, 189, 32–42. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | Reference |
|---|---|---|
| CXCL10 | F 5-ACCCAAGTGCTGCCGTCATT-3’ R 5-ATTCTCACTGGCCCGTCATC-3’ | [21] |
| Gad1 | F 5’-CGCTTGGCTTTGGAACCGACAA-3’ R 5’-GAATGCTCCGTAAACAGTCGTGC-3’ | NM_008077 |
| GFAP | F 5’-CGGGAGTCGGCCAGTTACCAG-3’ R 5’-TTTCCTGTAGGTGGCGATCTC-3’ | [21] |
| Map2 | F 5’-GCCAGCCTCGGAACAAACA-3’ R 5’-GCTCAGCGAATGAGGAAGGA-3’ | [22] |
| Reelin | F 5’-CCCAGCCCAGACAGACAGTT-3’ R 3’-CCAGGTGATGCCATTGTTGA-3’ | [23] |
| Parvalbumin | F 5’-TGTCGATGACAGACGTGCTC-3’ R 5’-TTCTTCAACCCCAATCTTGC-3’ | [24] |
| Somatostatin | F 5’-TCTGCATCGTCCTGGCTTT-3’ R 5’-CTTGGCCAGTTCCTGTTTCC-3’ | [25] |
| S12 | F 5’-GGCATAGCTGCTGGAGGTGTAA-3’ R 5’-GGGCTTGGCGCTTGTCTAA-3’ | [23] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Pozzo, V.; Crowell, B.; Briski, N.; Crockett, D.P.; D’Arcangelo, G. Reduced Reelin Expression in the Hippocampus after Traumatic Brain Injury. Biomolecules 2020, 10, 975. https://doi.org/10.3390/biom10070975
Dal Pozzo V, Crowell B, Briski N, Crockett DP, D’Arcangelo G. Reduced Reelin Expression in the Hippocampus after Traumatic Brain Injury. Biomolecules. 2020; 10(7):975. https://doi.org/10.3390/biom10070975
Chicago/Turabian StyleDal Pozzo, Valentina, Beth Crowell, Nicholas Briski, David P. Crockett, and Gabriella D’Arcangelo. 2020. "Reduced Reelin Expression in the Hippocampus after Traumatic Brain Injury" Biomolecules 10, no. 7: 975. https://doi.org/10.3390/biom10070975