Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Evolutionary Framework
2.3. Region Conservation
2.4. Positive Selection: Selectome and McDonald and Kreitman (MK) Test Results
3. Results
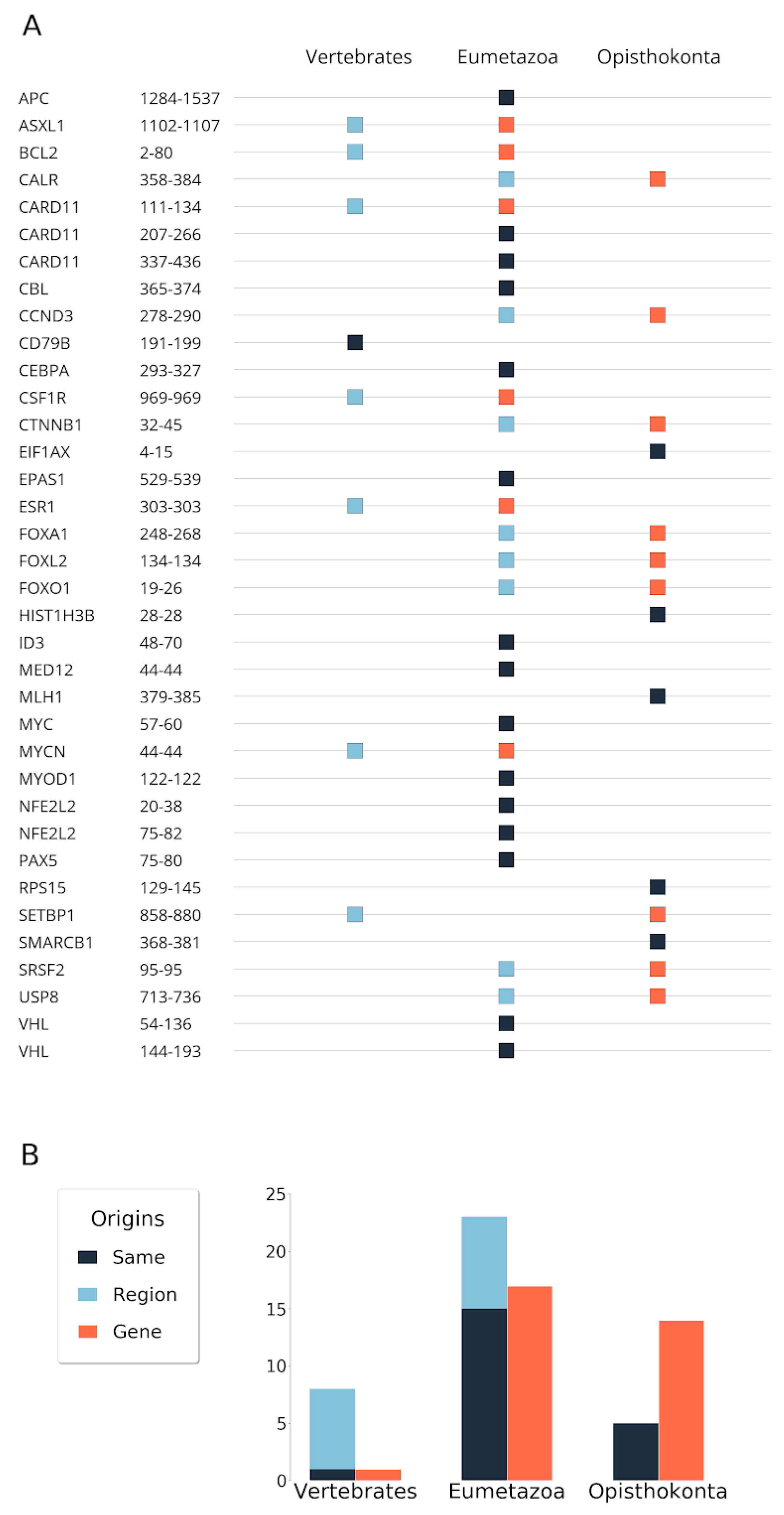
3.1. Evolutionary Origin of Genes and Regions
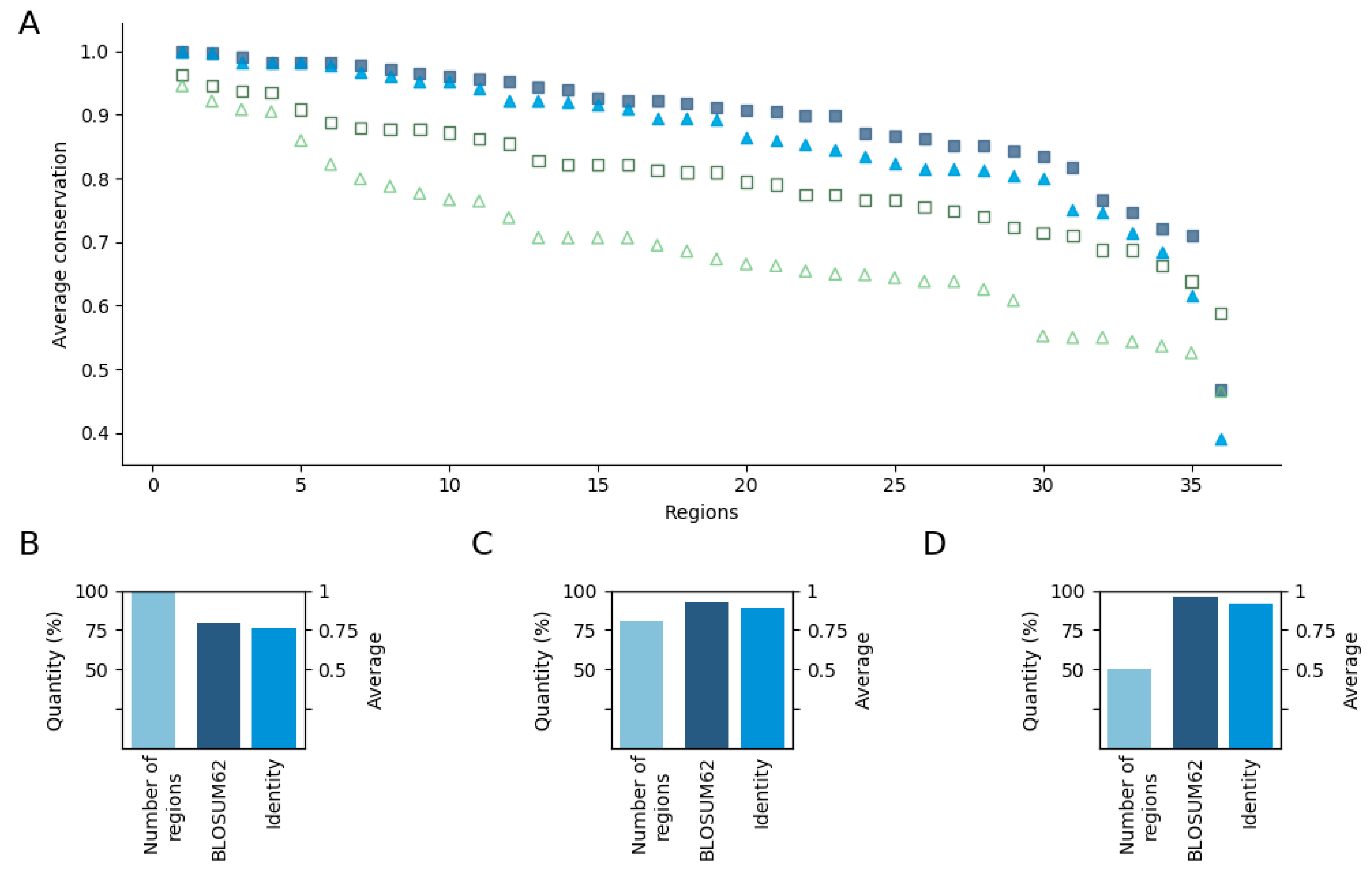
3.2. Position Conservation
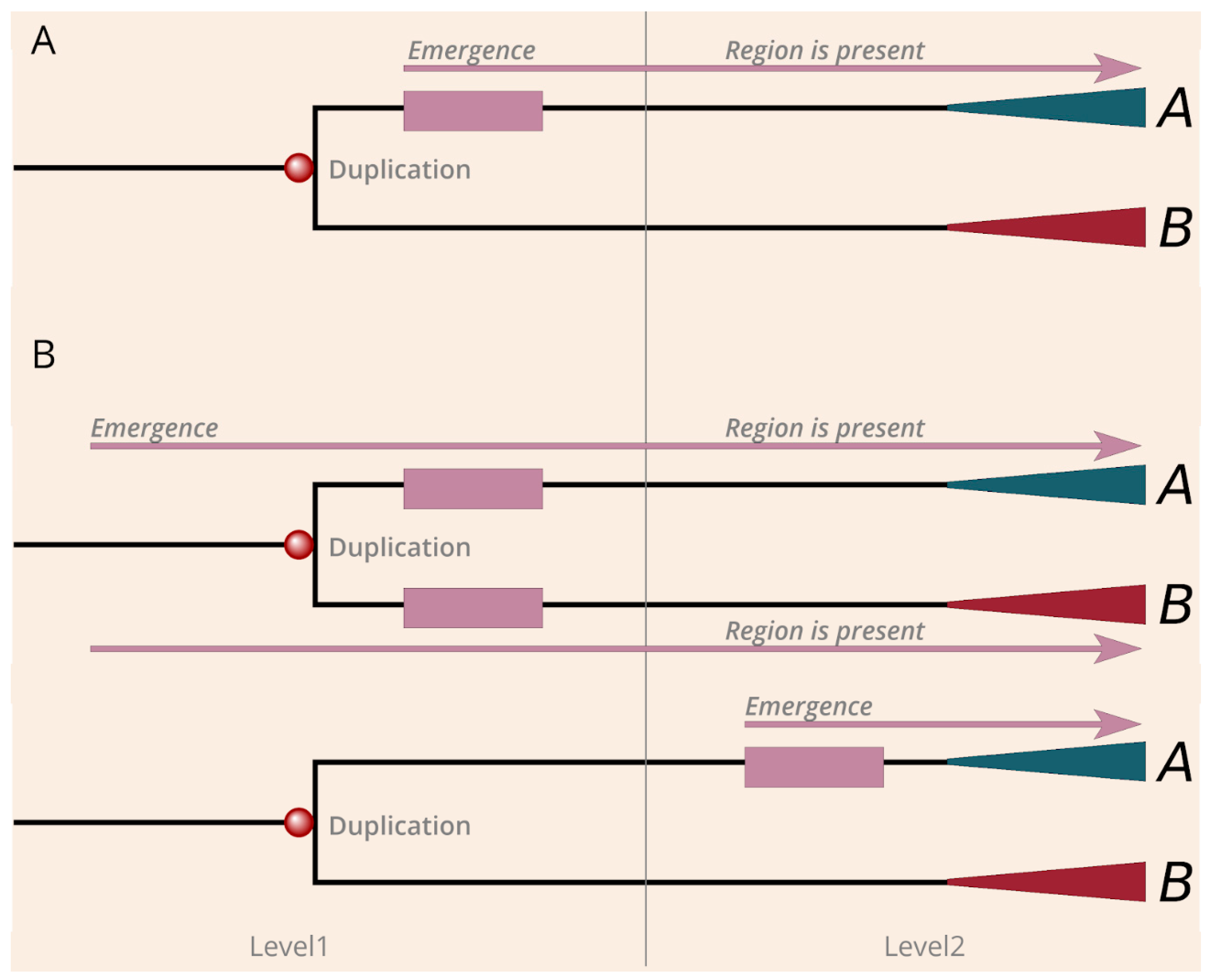
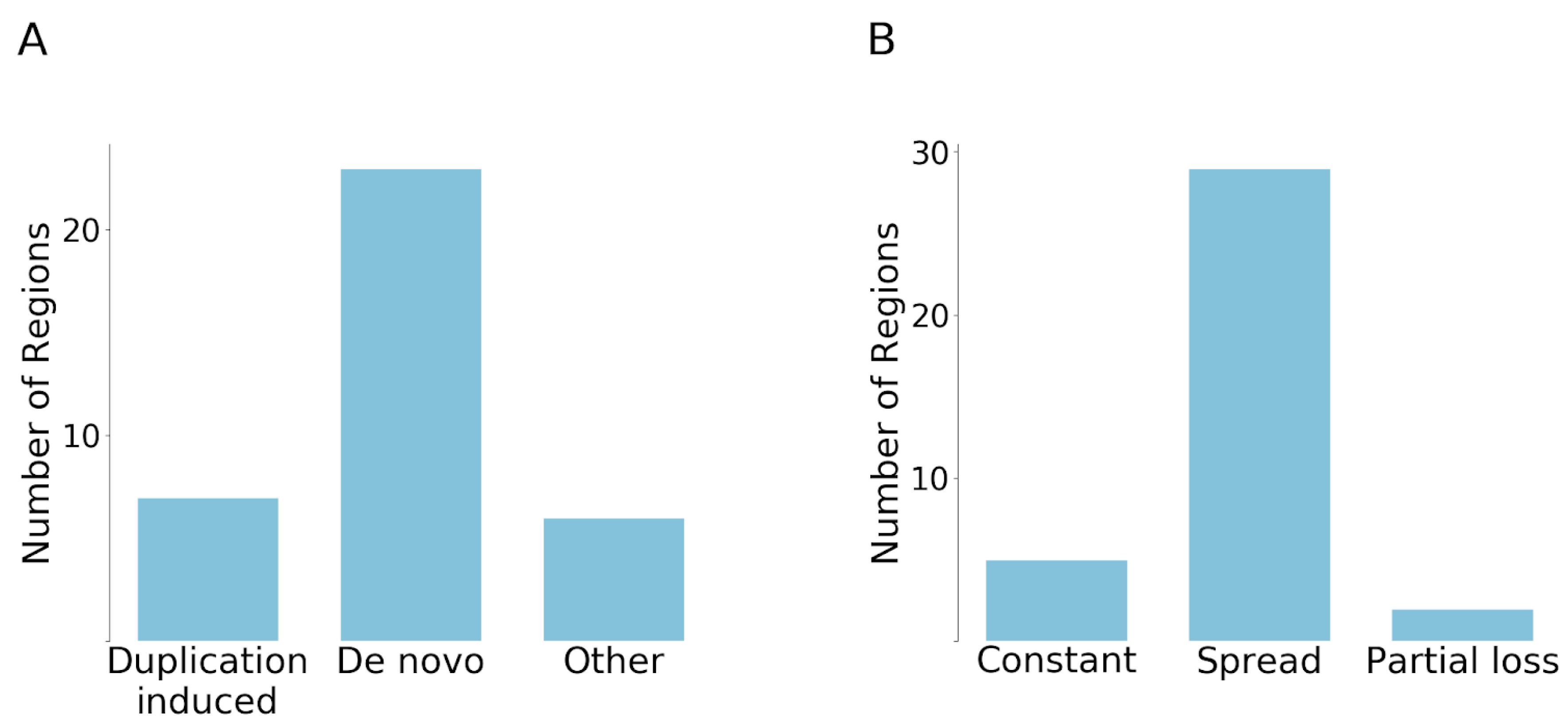
3.3. Contribution of Duplications to the Emergence of Disease Risk Regions
3.4. Case Studies
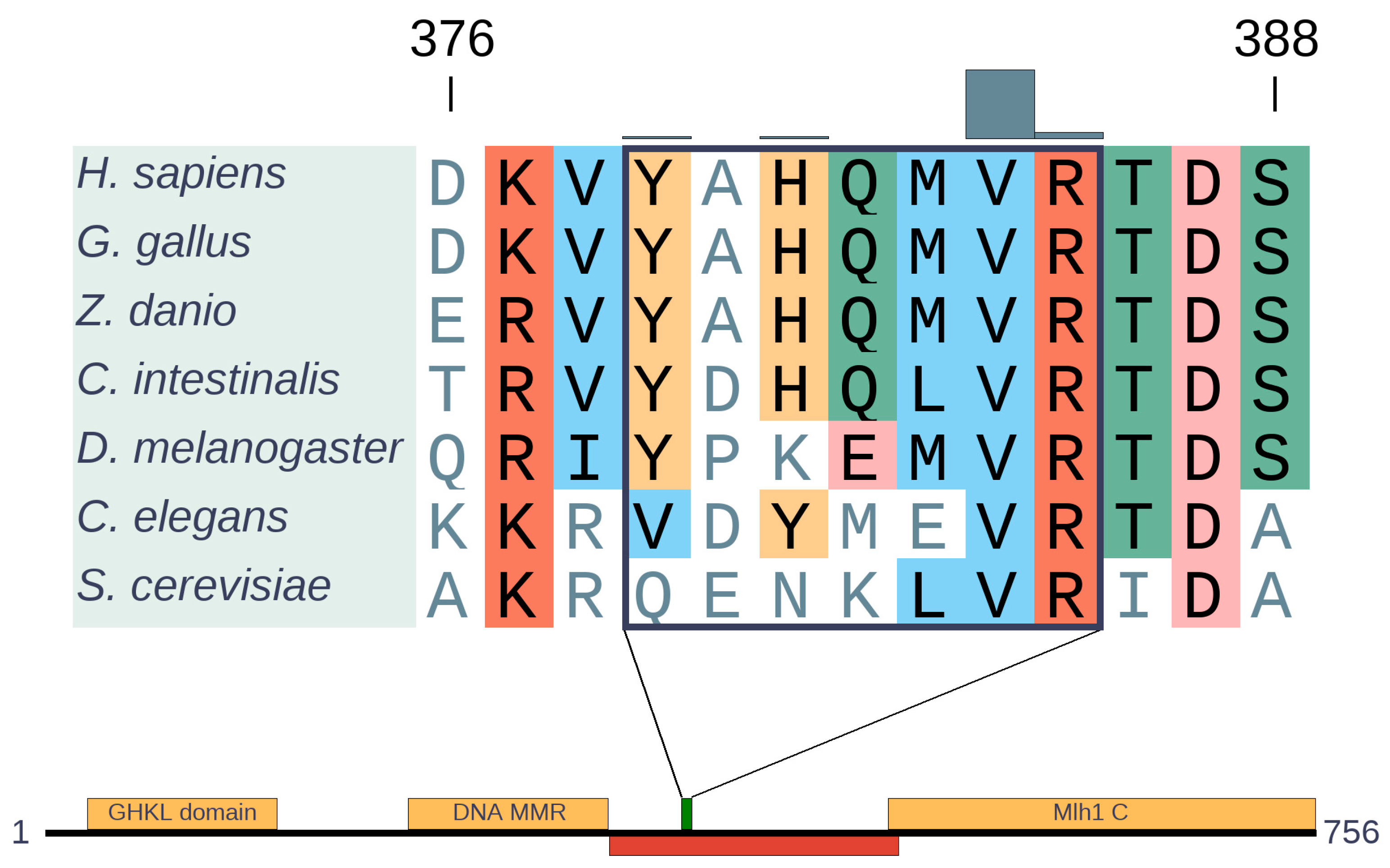
3.4.1. MLH1
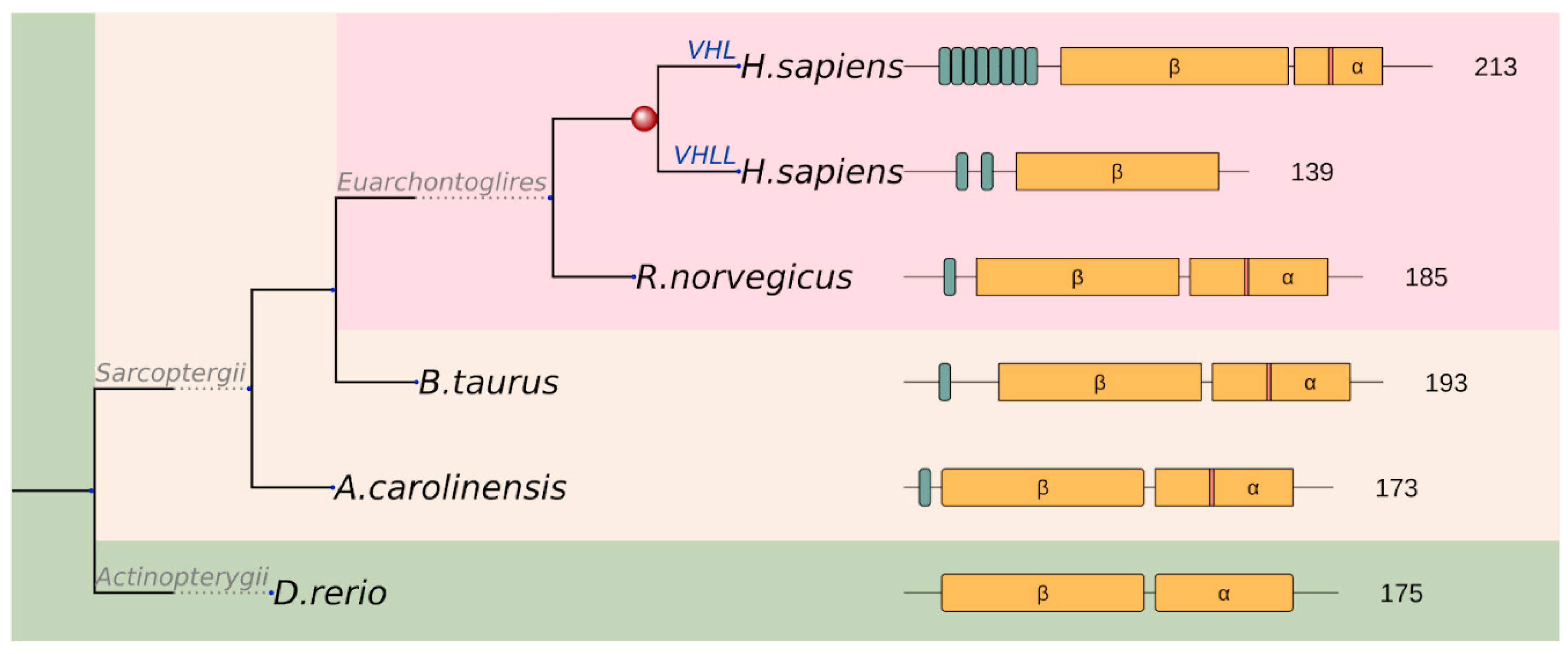
3.4.2. VHL
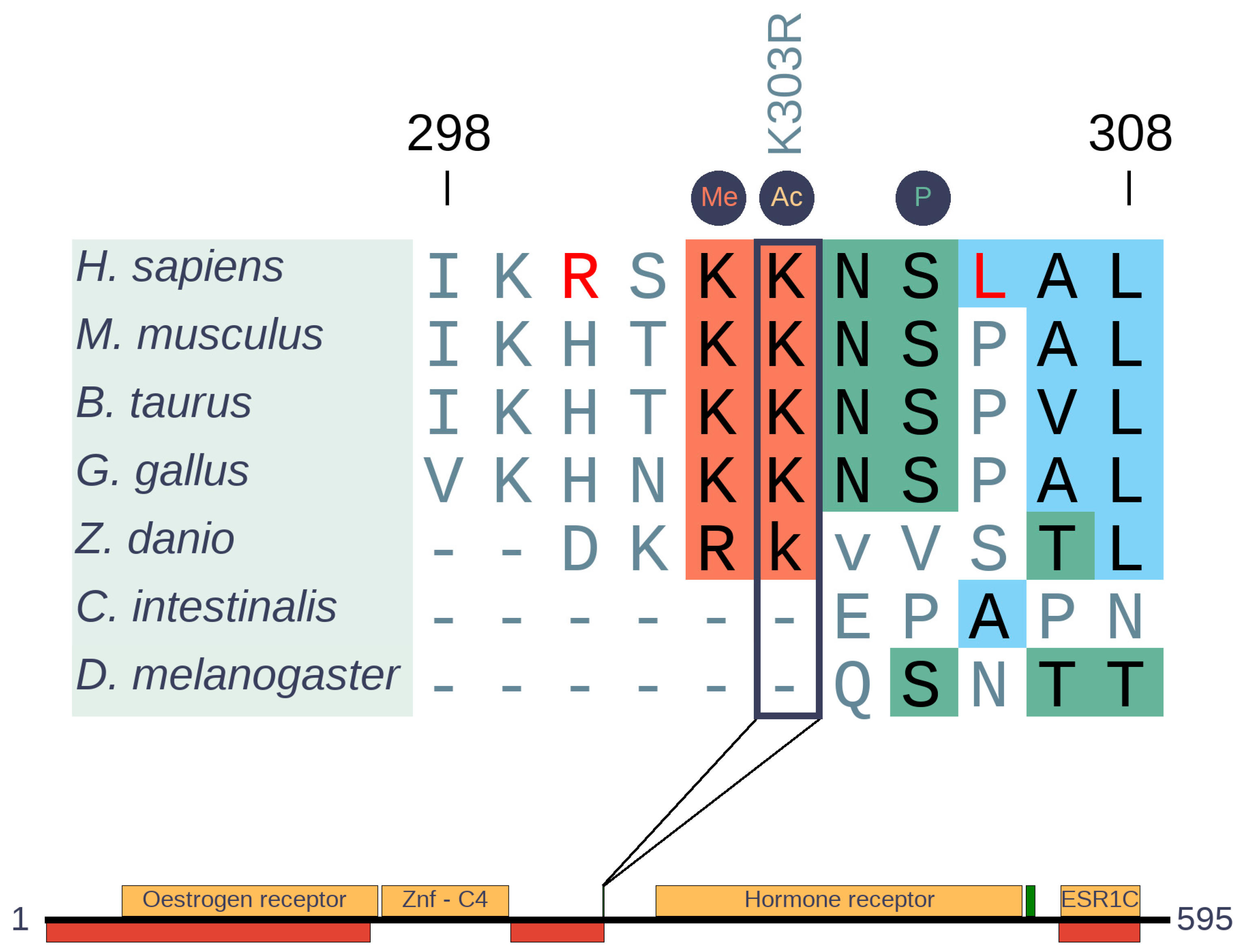
3.4.3. ESR1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jacob, F. Evolution and tinkering. Science 1977, 196, 1161–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef] [Green Version]
- Domazet-Loso, T.; Tautz, D. An ancient evolutionary origin of genes associated with human genetic diseases. Mol. Biol. Evol. 2008, 25, 2699–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickerson, J.E.; Robertson, D.L. On the origins of mendelian disease genes in man: The impact of gene duplication. Mol. Biol. Evol. 2012, 29, 2284. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Zeke, A.; Reményi, A.; Simon, I.; Dosztányi, Z. Systematic analysis of somatic mutations driving cancer: Uncovering functional protein regions in disease development. Biol. Direct 2016, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Hajdu-Soltész, B.; Zeke, A.; Dosztányi, Z. Intrinsically disordered protein mutations can drive cancer and their targeted interference extends therapeutic options. Bioinform. bioRxiv 2020, 2443. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446. [Google Scholar] [CrossRef]
- Davey, N.E.; Cyert, M.S.; Moses, A.M. Short linear motifs—Ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucleic Acids Res. 2011, 40, D84–D90. [Google Scholar] [CrossRef] [Green Version]
- Herrero, J.; Muffato, M.; Beal, K.; Fitzgerald, S.; Gordon, L.; Pignatelli, M.; Vilella, A.J.; Searle, S.M.J.; Amode, R.; Brent, S.; et al. Ensembl comparative genomics resources. Database 2016, 2016, bav096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebeskind, B.J.; McWhite, C.D.; Marcotte, E.M. Towards consensus gene ages. Genome Biol. Evol. 2016, 8, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Moretti, S.; Laurenczy, B.; Gharib, W.; Castella, B.; Kuzniar, A.; Schabauer, H.; Studer, R.A.; Valle, M.; Salamin, N.; Stockinger, H.; et al. Selectome update: Quality control and computational improvements to a database of positive selection. Nucleic Acids Res. 2013, 42, D917–D921. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Gayà-Vidal, M.; Alba, M.M. Uncovering adaptive evolution in the human lineage. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Berg, T.K.V.D.; Yoder, J.A.; Litman, G. On the origins of adaptive immunity: Innate immune receptors join the tale. Trends Immunol. 2004, 25, 11–16. [Google Scholar] [CrossRef]
- Wu, G.; Xu, G.; Schulman, B.A.; Jeffrey, P.D.; Harper, J.W.; Pavletich, N.P. Structure of a beta-TrCP1-Skp1-beta-catenin complex: Destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol. Cell 2003, 11, 1445–1456. [Google Scholar] [CrossRef]
- Shcherbakova, P.V.; Kunkel, T.A. Mutator Phenotypes conferred by MLH1 Overexpression and by Heterozygosity for mlh1 Mutations. Mol. Cell. Biol. 1999, 19, 3177–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Shimodaira, H.; Andreutti-Zaugg, C.; Iggo, R.; Kolodner, R.D.; Ishioka, C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair Assays. Cancer Res. 2007, 67, 4595–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akagi; Ohsawa, T.; Sahara, T.; Muramatsu, S.; Nishimura, Y.; Yathuoka, T.; Tanaka, Y.; Yamaguchi, K.; Ishida, H. Colorectal cancer susceptibility associated with the hMLH1 V384D variant. Mol. Med. Rep. 2009, 2, 887–891. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, H.S.; Kim, K.-Y.; Park, J.-H.; Roh, H.; Park, H.Y.; Kim, W.S. High prevalence of the MLH1 V384D germline mutation in patients with HER2-positive luminal B breast cancer. Sci. Rep. 2019, 9, 10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueneau, E.; Dhérin, C.; Legrand, P.; Tellier-Lebègue, C.; Gilquin, B.; Bonnesoeur, P.; Londino, F.; Quemener, C.; Le Du, M.-H.; A Márquez, J.; et al. Structure of the MutLα C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat. Struct. Mol. Biol. 2013, 20, 461–468. [Google Scholar] [CrossRef]
- Kim, Y.; Furman, C.M.; Manhart, C.M.; Alani, E.; Finkelstein, I.J. Intrinsically disordered regions regulate both catalytic and non-catalytic activities of the MutLα mismatch repair complex. Nucleic Acids Res. 2018, 47, 1823–1835. [Google Scholar] [CrossRef] [Green Version]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.W.; Nakayama, K.I. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef] [Green Version]
- Cardote, T.A.; Gadd, M.S.; Ciulli, A. Crystal structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase complex. Structure 2017, 25, 901–911.e3. [Google Scholar] [CrossRef] [Green Version]
- Sutovsky, H.; Gazit, E. The von Hippel-Lindau tumor suppressor protein is a molten globule under native conditions. J. Biol. Chem. 2004, 279, 17190–17196. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Verma, S.C.; Kumar, P.; Ma, M.; Robertson, E.S. Hypoxia Inactivates the VHL tumor suppressor through PIASy-Mediated SUMO modification. PLoS ONE 2010, 5, e9720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Robertson, E.S. Ubiquitin/SUMO modification regulates VHL protein stability and nucleocytoplasmic localization. PLoS ONE 2010, 5, e12636. [Google Scholar] [CrossRef] [PubMed]
- Minervini, G.; Mazzotta, G.; Masiero, A.; Sartori, E.; Corrà, S.; Potenza, E.; Costa, R.; Tosatto, S.C.E. Isoform-specific interactions of the von Hippel-Lindau tumor suppressor protein. Sci. Rep. 2015, 5, 12605. [Google Scholar] [CrossRef] [PubMed]
- German, P.; Bai, S.; Liu, X.-D.; Sun, M.; Zhou, L.; Kalra, S.; Zhang, X.; Minelli, R.; Scott, K.L.; Mills, G.B.; et al. Phosphorylation-dependent cleavage regulates von Hippel Lindau proteostasis and function. Oncogene 2016, 35, 4973–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Gervais, M.L.; Li, W.; DeCaprio, J.A.; Challis, J.R.G.; Ohh, M. Molecular cloning and characterization of the von Hippel-Lindau-like protein. Mol. Cancer Res. 2004, 2, 43–52. [Google Scholar]
- Dhayalan, A.; Kudithipudi, S.; Rathert, P.; Jeltsch, A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem. Biol. 2011, 18, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Fu, M.; Angeletti, R.H.; Siconolfi-Baez, L.; Reutens, A.T.; Albanese, C.; Lisanti, M.P.; Katzenellenbogen, B.S.; Kato, S.; Hopp, T.; et al. Direct Acetylation of the Estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J. Biol. Chem. 2001, 276, 18375–18383. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.-A.; Mazumdar, A.; Vadlamudi, R.K.; Kumar, R. P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-α and promotes hyperplasia in mammary epithelium. EMBO J. 2002, 21, 5437–5447. [Google Scholar] [CrossRef]
- Michalides, R.; Griekspoor, A.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; vant Veer, L.; Neefjes, J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell 2004, 5, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Zhang, M.; Pestell, R.; Curran, E.M.; Welshons, W.V.; Fuqua, S.A.W. Phosphorylation of estrogen receptor α blocks its Acetylation and regulates estrogen sensitivity. Cancer Res. 2004, 64, 9199–9208. [Google Scholar] [CrossRef] [Green Version]
- Rust, H.L.; Thompson, P.R. Kinase consensus sequences: A breeding ground for crosstalk. ACS Chem. Biol. 2011, 6, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leeuw, R.; Flach, K.; Toaldo, C.B.; Alexi, X.; Canisius, S.; Neefjes, J.; Michalides, R.; Zwart, W. PKA phosphorylation redirects ERα to promoters of a unique gene set to induce tamoxifen resistance. Oncogene 2012, 32, 3543–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsriku, C.; Britton, D.J.; Held, J.M.; Schilling, B.; Scott, G.K.; Gibson, B.W.; Benz, C.C.; Baldwin, M.A. Systematic mapping of posttranslational modifications in human estrogen receptor-α with emphasis on novel phosphorylation sites. Mol. Cell. Proteom. 2008, 8, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.C.; Basu, A.; El-Gharbawy, A.; Carrier, L.; Smith, C.L.; Rowan, B.G. Identification of four novel phosphorylation sites in estrogen receptor α: Impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. 2009, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Walker, V.R.; Korach, K. Estrogen receptor knockout mice as a model for endocrine research. ILAR J. 2004, 45, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porteous, R.; Herbison, A.E. Genetic deletion of Esr1 in the mouse preoptic area disrupts the LH surge and estrous cyclicity. Endocrinology 2019, 160, 1821–1829. [Google Scholar] [CrossRef]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Ellison, A.R.; Lofing, J.; Bitter, G.A. Human MutL homolog (MLH1) function in DNA mismatch repair: A prospective screen for missense mutations in the ATPase domain. Nucleic Acids Res. 2004, 32, 5321–5338. [Google Scholar] [CrossRef] [Green Version]
- Duchatel, R.J.; Jackson, E.R.; Alvaro, F.; Nixon, B.; Hondermarck, H.; Dun, M.D. Signal transduction in diffuse intrinsic Pontine Glioma. Proteomics 2019, 19, e1800479. [Google Scholar] [CrossRef] [Green Version]
- Piazza, R.; Magistroni, V.; Redaelli, S.; Mauri, M.; Massimino, L.; Sessa, A.; Peronaci, M.; Lalowski, M.M.; Soliymani, R.; Mezzatesta, C.; et al. SETBP1 induces transcription of a network of development genes by acting as an epigenetic hub. Nat. Commun. 2018, 9, 2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Marcos, P.; Zhou, F.; Karunasiri, C.; Zhang, F.; Dong, J.; Nanda, J.; Kulkarni, S.D.; Sen, N.D.; Tamame, M.; Zeschnigk, M.; et al. eIF1A residues implicated in cancer stabilize translation preinitiation complexes and favor suboptimal initiation sites in yeast. eLife 2017, 6, e31250. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Álvarez, M.G.; Arango, J.R.; Rodríguez, D.; Nadeu, F.; Prado, M.A.; Valdés-Mas, R.; Puente, D.A.; Paulo, J.A.; Delgado, J.; et al. Altered patterns of global protein synthesis and translational fidelity in RPS15-mutated chronic lymphocytic leukemia. Blood 2018, 132, 2375–2388. [Google Scholar] [CrossRef] [Green Version]
- Masaki, S.; Ikeda, S.; Hata, A.; Shiozawa, Y.; Kon, A.; Ogawa, S.; Suzuki, K.; Hakuno, F.; Takahashi, S.-I.; Kataoka, N. Myelodysplastic syndrome-associated SRSF2 mutations cause splicing changes by altering binding motif sequences. Front. Genet. 2019, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Assis, R.; Bachtrog, D. Rapid divergence and diversification of mammalian duplicate gene functions. BMC Evol. Biol. 2015, 15, 138. [Google Scholar] [CrossRef] [Green Version]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D. How the evolution of multicellularity set the stage for cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Positions under Positive Selection Referring to the Human Protein Sequence |
|---|---|
| CALR | 83(0.971), 155(0.971), 177(0.990), 267(0.995), 307(0.994), 336(0.991), 360(0.999) |
| CTNNB1 | 121(0.999), 206(0.993), 250(0.998), 287(0.991), 411(0.998), 433(0.993), 525(0.997), 552(0.998), 556(0.916) |
| VHL | 127(0.957), 132(0.942), 141(0.923), 171(0.947), 183(0.963), 185(0.920) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajkos, M.; Zeke, A.; Dosztányi, Z. Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions. Biomolecules 2020, 10, 1115. https://doi.org/10.3390/biom10081115
Pajkos M, Zeke A, Dosztányi Z. Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions. Biomolecules. 2020; 10(8):1115. https://doi.org/10.3390/biom10081115
Chicago/Turabian StylePajkos, Mátyás, András Zeke, and Zsuzsanna Dosztányi. 2020. "Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions" Biomolecules 10, no. 8: 1115. https://doi.org/10.3390/biom10081115
APA StylePajkos, M., Zeke, A., & Dosztányi, Z. (2020). Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions. Biomolecules, 10(8), 1115. https://doi.org/10.3390/biom10081115