Glycine in Water Favors the Polyproline II State




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.1.1. Simulations of Tripeptides in Water
2.1.2. Simulations of Triglycine in Dimethyl Sulfoxide
2.1.3. Simulations of Triglycine in Tetrachloride
2.2. Analysis
2.2.1. J-Coupling Constants, Amide I’ Profiles, Gaussian Model, and Functions
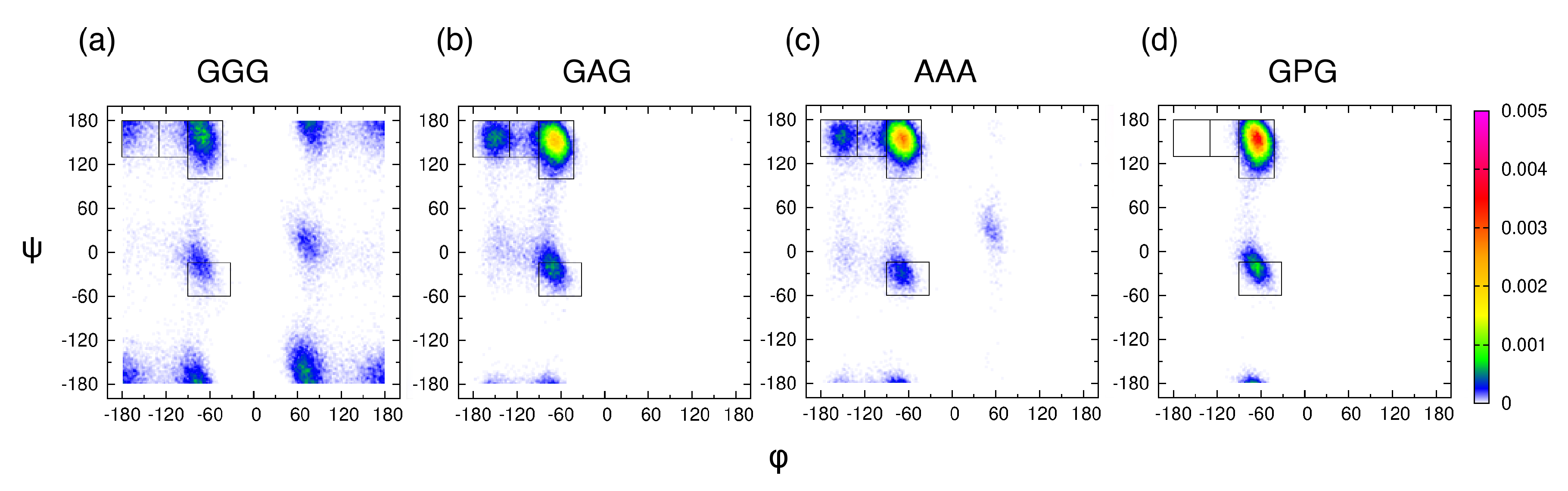
2.2.2. Definition of Mesostates
2.2.3. Shannon Entropy
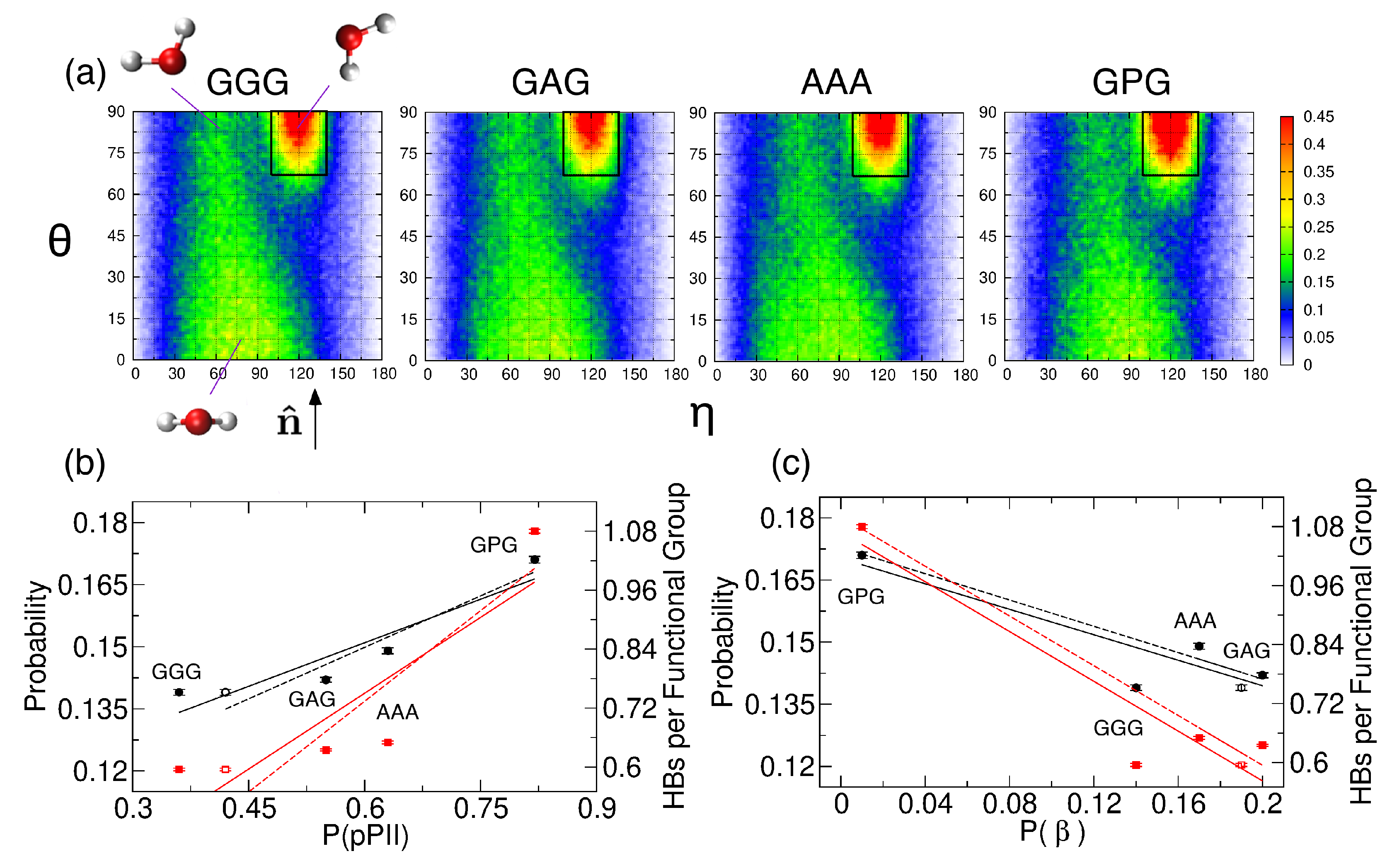
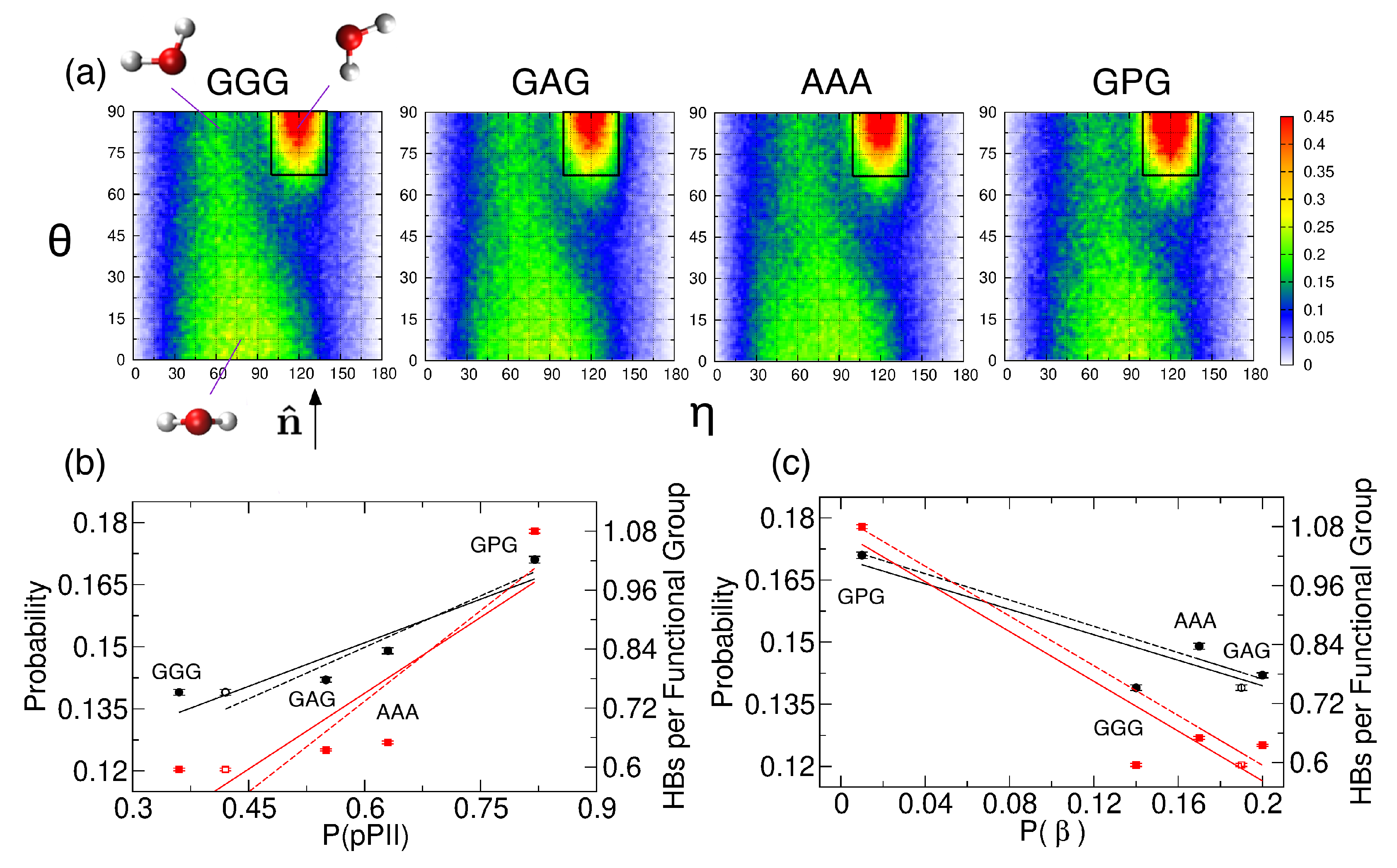
2.2.4. Water Orientation Plots
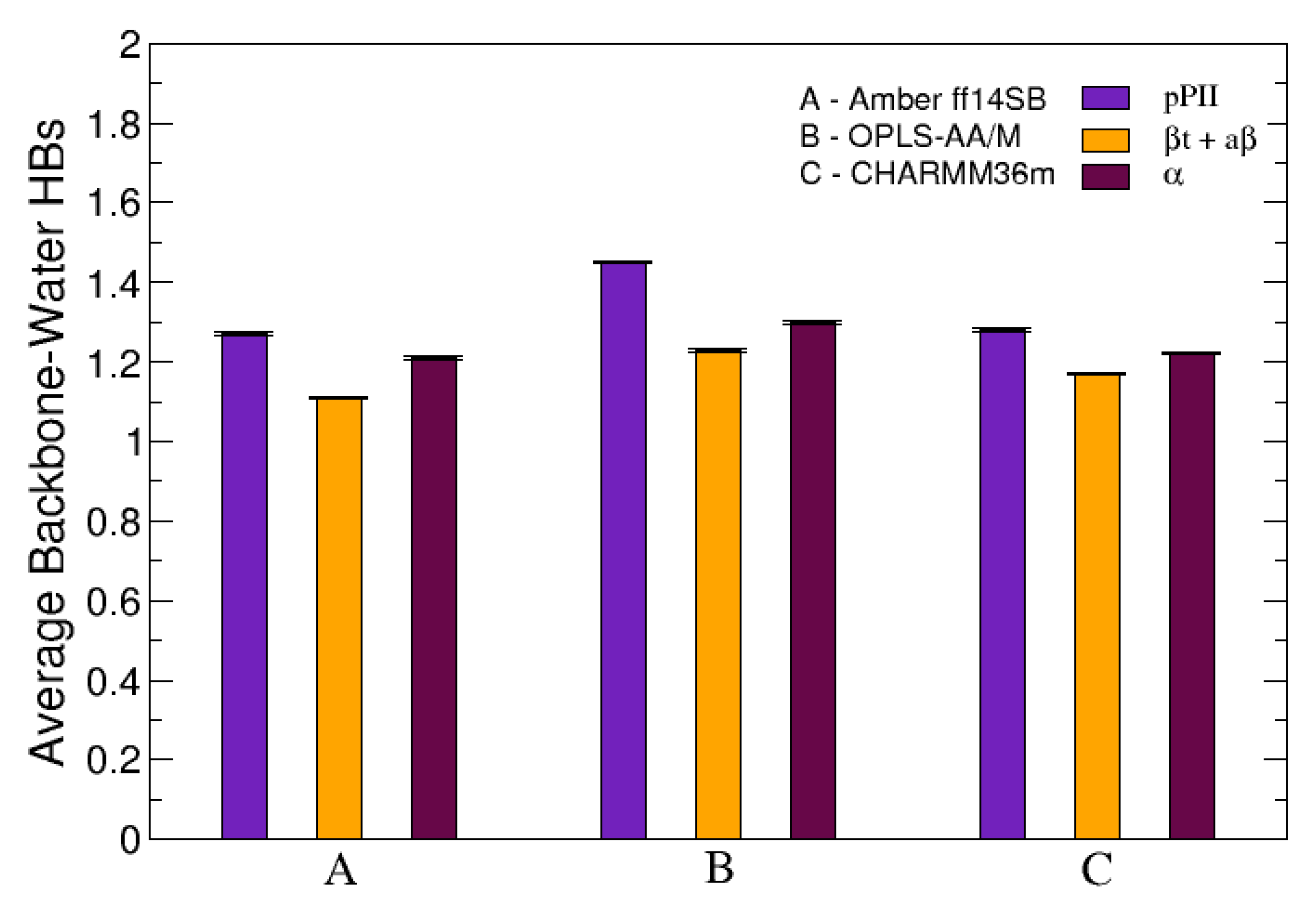
2.2.5. Solvent-Peptide and Intrapeptide HBs
3. Results
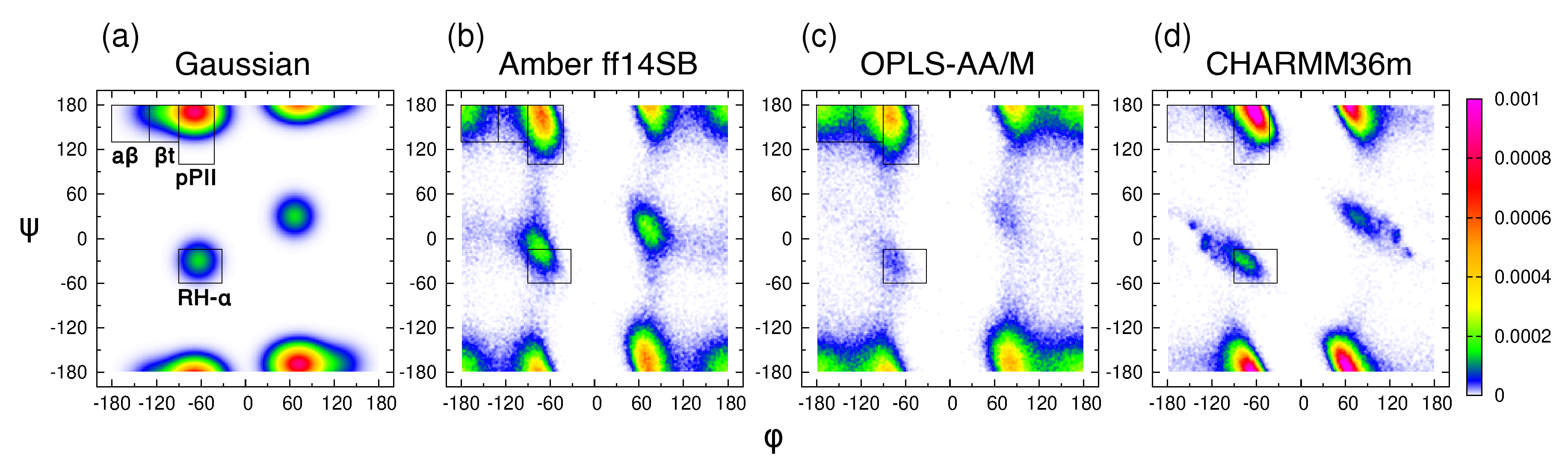
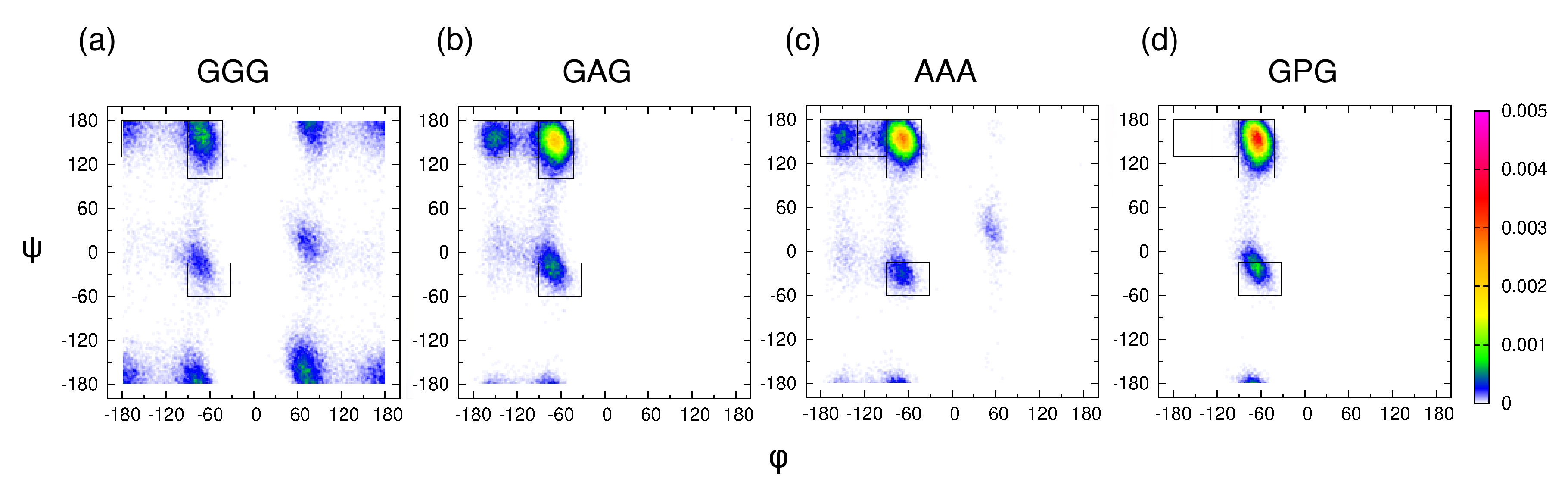
3.1. Experiment-Based Ramachandran Distribution for the Central Glycine in GGG Reveals High PPII Content
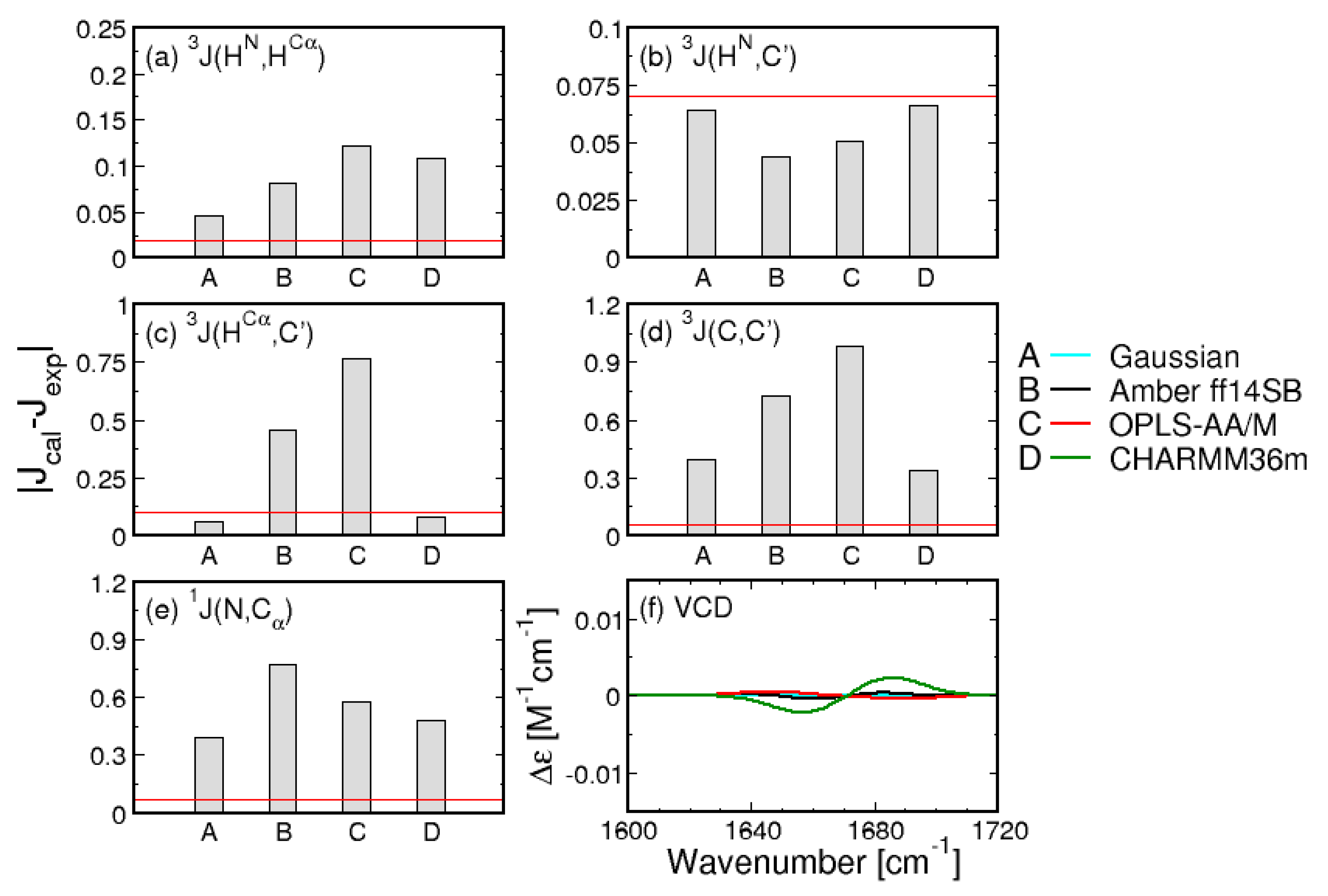
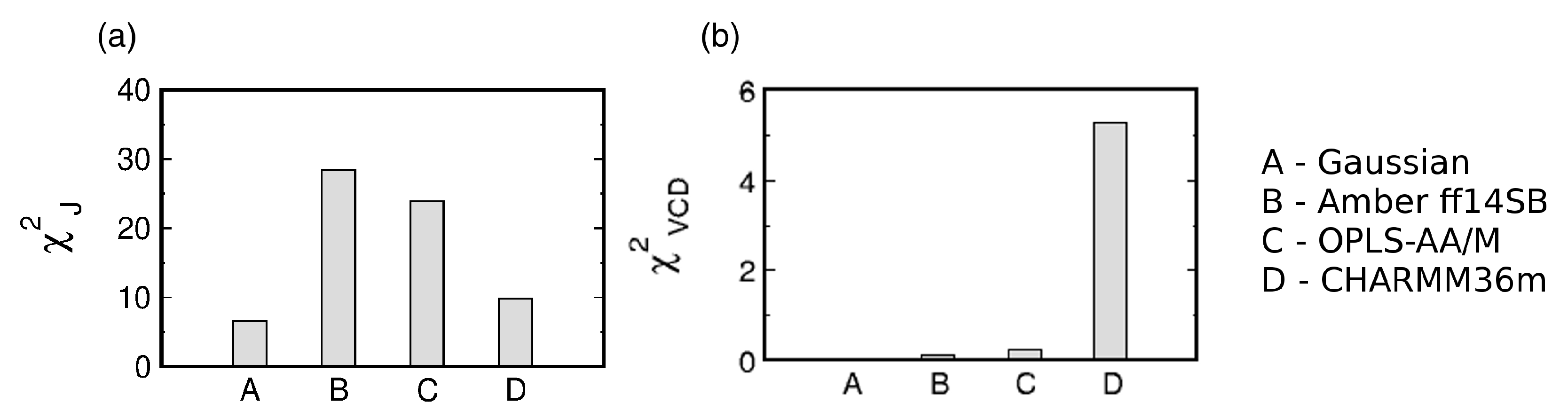
3.2. Conformational Ensembles of the Central Glycine in GGG: Assessment of MD Force Fields
3.3. The PPII State Enables Glycine and Alanine Residues to Form the Most HBs with Water
3.4. DMSO Reduces PPII Content of the Central Glycine in GGG
3.5. Nonpolar Solvent Further Reduces PPII Content of the Central Glycine in GGG
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| SAS | Solvent Accessible Surface |
| HB | Hydrogen Bond |
| pPII | polyproline II |
References
- Ramachandran, G.N.; Ramachandran, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95. [Google Scholar] [CrossRef]
- Ramakrishnan, C.; Ramachandran, G.N. Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units. Biophys. J. 1965, 5, 909–933. [Google Scholar] [CrossRef] [Green Version]
- Brant, D.A.; Flory, P.J.J. The Configuration of Random Polypeptide Chains. II. Theory. J. Am. Chem. Soc. 1965, 87, 2791–2800. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Mechanics of Chain Molecues; Wiley & Sons: New York, NY, USA, 1969; pp. 30–31. [Google Scholar]
- Kauzmann, W. Some factors in the interpretation of protein denaturation. Adv. Prot. Chem. 1959, 14, 1–63. [Google Scholar]
- Lyle, N.; Das, R.K.; Pappu, R.V. A quantitative measure for protein conformational heterogeneity. J. Chem. Phys. 2013, 139, 121907. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Garai, K.; Lyle, N.; Vitalis, A.; Pappu, R.V. Quantitative Assessments of the Distinct Contributions of Polypeptide Backbone Amides versus Side Chain Groups to Chain Expansion via Chemical Denaturation. J. Am. Chem. Soc. 2015, 137, 2984–2995. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.K.; Brasseur, R. The Ramachandran plots of glycine and pre-proline. BMC Struct. Biol 2005, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.S.; Chen, K.; Liu, Z.G.; Kallenbach, N.R. Conformation of the backbone in unfolded proteins. Chem. Rev. 2006, 106, 1877–1897. [Google Scholar] [CrossRef]
- Toal, S.E.; Schweitzer-Stenner, R. Local order in the unfolded state: Conformational biases and nearest neighbor interactions. Biomolecules 2014, 4, 725–773. [Google Scholar] [CrossRef]
- Avbelj, F.; Grdadolnik, S.G.; Grdadolnik, J.; Baldwin, R.L. Intrinsic backbone preferences are fully present in blocked amino acids. Proc. Natl. Acad. Sci. USA 2006, 103, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Colubri, A.; Zaman, M.H.; Koide, S.; Sosnick, T.R.; Freed, K.F. Helix, sheet, and polyproline II frequencies and strong nearest neighbor effects in a restricted coil library. Biochemistry 2005, 44, 9691–9702. [Google Scholar] [CrossRef] [Green Version]
- DeBartolo, J.; Jha, A.; Freed, K.F.; Sosnick, T.R. Ch.3: Local Backbone Preferences and Nearest-Neighbor Effects in the Unfolded and Native States. In Folding, Misfolding and Nonfolding of Peptides and Small Proteins; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Grdadolnik, J.; Grdadolnik, S.G.; Avbelj, F. Determination of conformational preferences of dipeptides using vibrational spectroscopy. J. Phys. Chem. B 2008, 112, 2712–2718. [Google Scholar] [CrossRef]
- Makowska, J.; Rodziewicz-Motowidlo, S.; Baginska, K.; Vila, J.A.; Liwo, A.; Chmurzynski, L.; Scheraga, H.A. Polyproline II conformation is one of many local conformational states and is not an overall conformation of unfolded peptides and proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 1744–1749. [Google Scholar] [CrossRef] [Green Version]
- Makowska, J.; Rodziewicz-Motowidlo, S.; Baginska, K.; Makowski, M.; Vila, J.A.; Liwo, A.; Chmurzynski, L.; Scheraga, H.A. Further evidence for the absence of polyproline II stretch in the XAO peptide. Biophys. J. 2007, 92, 2904–2917. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.S.; Woody, R.W.; Kallenbach, N.R. Is polyproline II a major backbone conformation in unfolded proteins? Adv. Prot. Chem. 2002, 62, 163–240. [Google Scholar]
- Graf, J.; Nguyen, P.H.; Stock, G.; Schwalbe, H. Structure and dynamics of the homologous series of alanine peptides: A joint molecular dynamics/NMR study. J. Am. Chem. Soc. 2007, 129, 1179–1189. [Google Scholar] [CrossRef]
- Hagarman, A.; Measey, T.J.; Mathieu, D.; Schwalbe, H.; Schweitzer-Stenner, R. Intrinsic Propensities of Amino Acid Residues in GxG Peptides Inferred from Amide I’ Band Profiles and NMR Scalar Coupling Constants. J. Am. Chem. Soc. 2010, 132, 540–551. [Google Scholar] [CrossRef]
- Toal, S.; Meral, D.; Verbaro, D.; Urbanc, B.; Schweitzer-Stenner, R. The pH-independence of trialanine and the effects of termini blocking in short peptides: A combined vibrational, NMR, UVCD, and molecular dynamics study. J. Phys. Chem. B 2013, 117, 3689–3706. [Google Scholar] [CrossRef] [Green Version]
- Zaman, M.H.; Shen, M.Y.; Berry, R.S.; Freed, K.F.; Sosnick, T.R. Investigations into sequence and conformational dependence of backbone entropy, inter-basin dynamics and the Flory isolated-pair hypothesis for peptides. J. Mol. Biol. 2003, 331, 693–711. [Google Scholar] [CrossRef]
- Hagarman, A.; Mathieu, D.; Toal, S.; Measey, T.J.; Schwalbe, H.; Schweitzer-Stenner, R. Amino Acids with Hydrogen-Bonding Side Chains have an Intrinsic Tendency to Sample Various Turn Conformations in Aqueous Solution. Chemistry 2011, 17, 6789–6797. [Google Scholar] [CrossRef]
- Rybka, K.; Toal, S.E.; Verbaro, D.J.; Mathieu, D.; Schwalbe, H.; Schweitzer-Stenner, R. Disorder and order in unfolded and disordered peptides and proteins: A view derived from tripeptide conformational analysis. II. Tripeptides with short side chains populating asx and β-type like turn conformations. Proteins Struct. Funct. Bioinf. 2013, 81, 968–983. [Google Scholar] [CrossRef] [PubMed]
- Abascal, J.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef] [PubMed]
- Meral, D.; Toal, S.; Schweitzer-Stenner, R.; Urbanc, B. Water-centered interpretation of intrinsic pPII propensities of amino acid residues: In vitro-driven molecular dynamics study. J. Phys. Chem. B 2015, 119, 13237–13251. [Google Scholar] [CrossRef] [PubMed]
- Baxa, M.C.; Haddadian, E.J.; Jha, A.K.; Freed, K.F.; Sosnick, T.R. Context and force field dependence of the loss of protein backbone entropy upon folding using realistic denatured and native state ensembles. J. Am. Chem. Soc. 2012, 134, 15929–15936. [Google Scholar] [CrossRef] [Green Version]
- Avbelj, F.; Baldwin, R.L. Role of backbone solvation and electrostatics in generating preferred peptide backbone conformations: Distributions of phi. Proc. Natl. Acad. Sci. USA 2003, 100, 5742–5747. [Google Scholar] [CrossRef] [Green Version]
- Avbelj, F.; Baldwin, R.L. Origin of the neighboring residue effect on peptide backbone conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 10967–10972. [Google Scholar] [CrossRef] [Green Version]
- Drozdov, A.; Grossfield, A.; Pappu, R. Role of solvent in determining conformational preferences of alanine dipeptide in water. J. Am. Chem. Soc. 2004, 126, 2574–2581. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Adrendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Genet. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Avbelj, F.; Baldwin, R.L. Limited validity of group additivity for the folding energetics of the peptide group. Proteins Struct. Funct. Bioinf. 2006, 63, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Naim, A. Molecular Theory of Water and Aqueous Solutions—Part II: The Role of Water in Protein Folding, Self-Assembly and Molecular Recognition; World Scientific: Singapore, 2011. [Google Scholar]
- König, G.; Bruckner, S.; Boresch, S. Absolute hydration free energies of blocked amino acids: Implications for protein solvation and stability. Biophys. J. 2013, 104, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer-Stenner, R.; Eker, F.; Huang, Q.; Griebenow, K. Dihedral angles of trialanine in D2O determined by combining FTIR and polarized visible Raman spectroscopy. J. Am. Chem. Soc. 2001, 123, 9628–9633. [Google Scholar] [CrossRef]
- Zhang, S.; Schweitzer-Stenner, R.; Urbanc, B. Do molecular dynamics force fields capture conformational dynamics of alanine in water? J. Chem. Theory Comput. 2020, 16, 510–527. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–364. [Google Scholar]
- Torii, H.; Tasumi, M. Model calculations on the amide-I infrared bands of globular proteins. J. Chem. Phys. 1992, 96, 3379–3387. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R. Secondary structure analysis of polypeptides based on an excitonic coupling model to describe the band profile of amide I ‘ of IR, Raman, and vibrational circular dichroism spectra. J. Phys. Chem. B 2004, 108, 16965–16975. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Pall, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Solving Software Challenges for Exascale, Lecture Notes in Computer Science, Proceedings of the 2nd International Conference on Exascale Applications and Software (EASC), Stockholm, Sweden, 2–3 April 2014; Markidis, S., Laure, E., Eds.; Cray Inc: Seattle, WA, USA; Mellanox Technologies: Sunnyvale, CA, USA, 2015; Volume 8759, pp. 3–27. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pall, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 8, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- da Silva Alan, S.; Wim, V. ACPYPE-Antechamber Python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- NIST Computational Chemistry Comparison and Benchmark Database. Available online: https://cccbdb.nist.gov/ (accessed on 23 June 2020).
- Schweitzer-Stenner, R. Distribution of Conformations Sampled by the Central Amino Acid Residue in Tripeptides Inferred From Amide I’ Band Profiles and NMR Scalar Coupling Constants. J. Phys. Chem. B 2009, 113, 2922–2932. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.C.; Bax, A. Determination of the backbone dihedral angles ϕ in human ubiquitin from reparametrized empirical Karplus equations. J. Am. Chem. Soc. 1996, 118, 2483–2494. [Google Scholar] [CrossRef]
- Hu, J.S.; Bax, A. Determination of ϕ and χ1 angles in proteins from 13C–13C three-bond J couplings measured by three-dimensional heteronuclear NMR. How planar is the peptide bond? J. Am. Chem. Soc. 1997, 119, 6360–6368. [Google Scholar] [CrossRef]
- Wirmer, J.; Schwalbe, H. Angular dependence of 1J(Ni, Cαi) and 2J(Ni, Cα(i-1)) coupling constants measured in J-modulated HSQCs. J. Biomol. NMR 2002, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.Y.; Gronenborn, A.M. Protein backbone 1HN–13Cα and 15N–13Cα residual dipolar and J couplings: New constraints for NMR structure determination. J. Am. Chem. Soc. 2004, 126, 6232–6233. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer-Stenner, R. Dihedral angles of tripeptides in solution directly determined by polarized Raman and FTIR spectroscopy. Biophys. J. 2002, 83, 523–532. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004, 126, 698–699. [Google Scholar] [CrossRef]
- Baruch-Shpigler, Y.; Wang, H.; Tuvi-Arad, I.; Avnir, D. Chiral Ramachandran plots I: Glycine. Biochemistry 2017, 56, 5635–5643. [Google Scholar] [PubMed]
- DiGuiseppi, D.; Milorey, B.; Lewis, G.; Kubatova, N.; Farrell, S.; Schwalbe, H.; Schweitzer-Stenner, R. Probing the conformation-dependent preferential binding of ethanol to cationic glycylalanylglycine in water/ethanol by vibrational and NMR spectroscopy. J. Phys. Chem. B 2017, 121, 5744–5758. [Google Scholar] [CrossRef]
- Toal, S.; Amidi, O.; Schweitzer-Stenner, R. Conformational Changes of Trialanine Induced by Direct Interactions between Alanine Residues and Alcohols in Binary Mixtures of Water with Glycerol and Ethanol. J. Am. Chem. Soc. 2011, 133, 12728–12739. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.T.R.; Cheng, R.P.; Wright, W.W.; Alonso, D.O.V.; Daggett, V.; Vanderkooi, J.M.; DeGrado, W.F. The hydration of amides in helices; a comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 2003, 12, 520–531. [Google Scholar] [CrossRef]
- Eker, F.; Cao, X.L.; Nafie, L.; Huang, Q.; Schweitzer-Stenner, R. The structure of alanine based tripeptides in water and dimethyl sulfoxide probed by vibrational spectroscopy. J. Phys. Chem. B 2003, 107, 358–365. [Google Scholar] [CrossRef]
- Milorey, B.; Farrell, S.; Toal, S.E.; Schweitzer-Stenner, R. Demixing of water and ethanol causes conformational redistribution and gelation of the cationic GAG tripeptide. Chem. Commun. 2015, 51, 16498–16501. [Google Scholar] [CrossRef]
- Gnanakaran, S.; Garcia, A. Validation of an all-atom protein force field: From dipeptides to larger peptides. J. Phys. Chem. B 2003, 107, 12555–12557. [Google Scholar] [CrossRef]
- Garcia, A.E. Characterization of non-α helical conformations in Ala peptides. Polymer 2004, 45, 669–676. [Google Scholar] [CrossRef]
- Tran, H.; Wang, X.; Pappu, R. Reconciling observations of sequence-specific conformational propensities with the generic polymeric behavior of denatured proteins. Biochemistry 2005, 44, 11369–11380. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.; Fitzkee, N.; Mezei, M.; Srinivasan, R.; Rose, G. A novel method reveals that solvent water favors polyproline II over β-strand conformation in peptides and unfolded proteins: Conditional hydrophobic accessible surface area (CHASA). Prot. Sci. 2005, 14, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilawe, N.V.; Raeber, A.E.; Schweitzer-Stenner, R.; Toal, S.E.; Wong, B.M. Assessing backbone solvation effects in the conformational propensities of amino acid residues in unfolded peptides. Phys. Chem. Chem. Phys. 2015, 17, 24917–24924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avbelj, F. Solvation and Electrostatics as Determinants of Local Structural Order in Unfolded Peptides and Proteins. In Protein and Peptide Folding, Misfolding, and Non-Folding; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2012; Chapter 5; pp. 131–158. [Google Scholar]
- Poon, C.D.; Samulski, E.T.; Weise, C.F.; Weisshaar, J.C. Do bridging water molecules dictate the structure of a model dipeptide in aqueous solution? J. Am. Chem. Soc. 2000, 122, 5642–5643. [Google Scholar] [CrossRef]
- Kentsis, A.; Mezei, M.; Osman, R. Origin of the sequence-dependent polyproline II structure in unfolded peptides. Proteins Struct. Funct. Bioinf. 2005, 61, 769–776. [Google Scholar] [CrossRef]
- Mezei, M.; Fleming, P.J.; Srinivasan, R.; Rose, G.D. Polyproline II helix is the preferred conformation for unfolded polyalanine in water. Proteins Struct. Funct. Bioinf. 2004, 55, 502–507. [Google Scholar] [CrossRef]
- Ben-Naim, A. Molecular Theory of Water and Aqueous Solutions—Part I: Understanding Water; World Scientific: Singapore, 2009. [Google Scholar]
- Toal, S.E.; Verbaro, D.J.; Schweitzer-Stenner, R. Role of enthalpy-entropy compensation interactions in determining the conformational propensities of amino acid residues in unfolded peptides. J. Phys. Chem. B 2014, 118, 1309–1318. [Google Scholar] [CrossRef]
- Lanza, G.; Chiacchio, M.A. Effects of hydration on the zwitterion trialanine conformation by electronic structure theory. J. Phys. Chem. B 2016, 120, 11705–11719. [Google Scholar] [CrossRef]
- Crick, F.H.; Rich, A. Structure of polyglycine II. Nature 1955, 176, 780–781. [Google Scholar] [CrossRef]
- Bykov, S.; Asher, S. Raman studies of solution polyglycine conformations. J. Phys. Chem. B 2010, 114, 6636–6641. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, A.A.; Sternberg, M.J.E.; Makarov, A.A. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef]
- Gates, Z.P.; Baxa, M.C.; Yu, W.; Riback, J.A.; Li, H.; Roux, B.; Kent, S.B.H.; Sosnick, T.R. Perplexing cooperative folding and stability of a low-sequence complexity, polyproline 2 protein lacking a hydrophobic core. Proc. Natl. Acad. Sci. USA 2017, 114, 2241–2246. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrews, B.; Zhang, S.; Schweitzer-Stenner, R.; Urbanc, B. Glycine in Water Favors the Polyproline II State. Biomolecules 2020, 10, 1121. https://doi.org/10.3390/biom10081121
Andrews B, Zhang S, Schweitzer-Stenner R, Urbanc B. Glycine in Water Favors the Polyproline II State. Biomolecules. 2020; 10(8):1121. https://doi.org/10.3390/biom10081121
Chicago/Turabian StyleAndrews, Brian, Shuting Zhang, Reinhard Schweitzer-Stenner, and Brigita Urbanc. 2020. "Glycine in Water Favors the Polyproline II State" Biomolecules 10, no. 8: 1121. https://doi.org/10.3390/biom10081121
APA StyleAndrews, B., Zhang, S., Schweitzer-Stenner, R., & Urbanc, B. (2020). Glycine in Water Favors the Polyproline II State. Biomolecules, 10(8), 1121. https://doi.org/10.3390/biom10081121