Tryptophanyl-tRNA Synthetase 1 Signals Activate TREM-1 via TLR2 and TLR4


Abstract
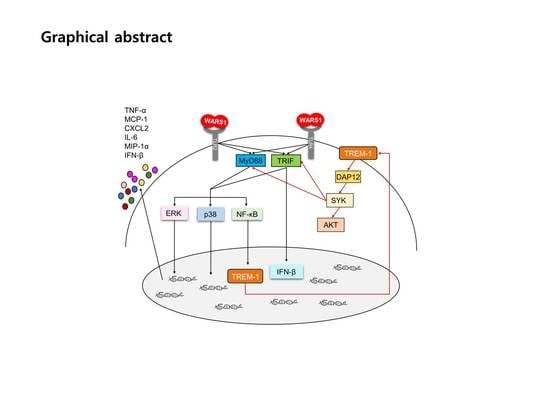
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Microarray Screening
2.2. Antibodies and Chemicals
2.3. Cell Culture
2.4. Recombinant Protein Purification
2.5. RNA Interference
2.6. GST Pulldown Assay
2.7. Quantification of RNA
2.8. Flow Cytometry
2.9. Immunofluorescence Assay
2.10. ELISA
2.11. WST Assay
2.12. Statistical Analysis
3. Results
3.1. WARS1 Mainly Activated Innate Inflammatory Response-Related Gene Expression in Human PBMCs
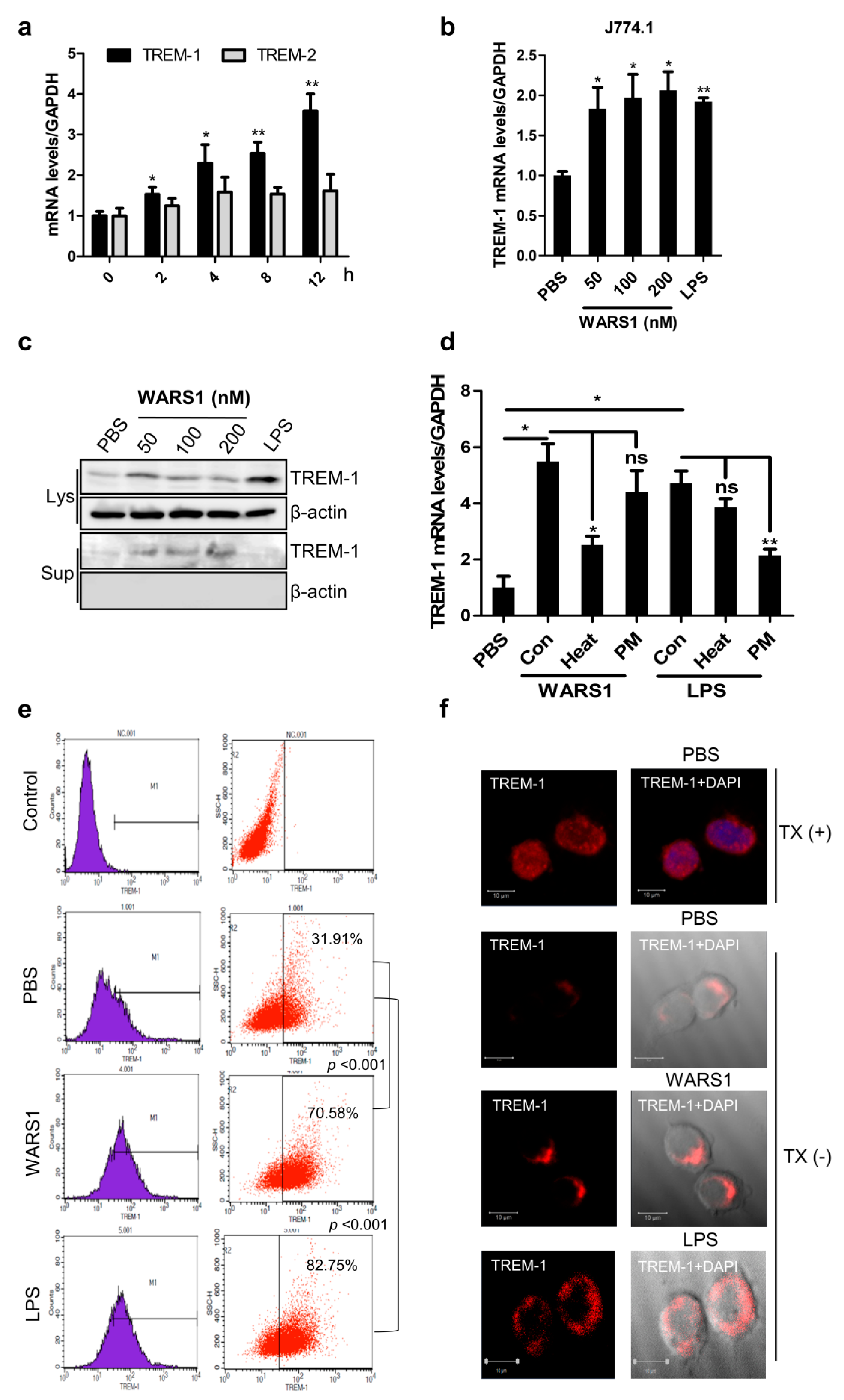
3.2. WARS1 Activated TREM-1 Expression
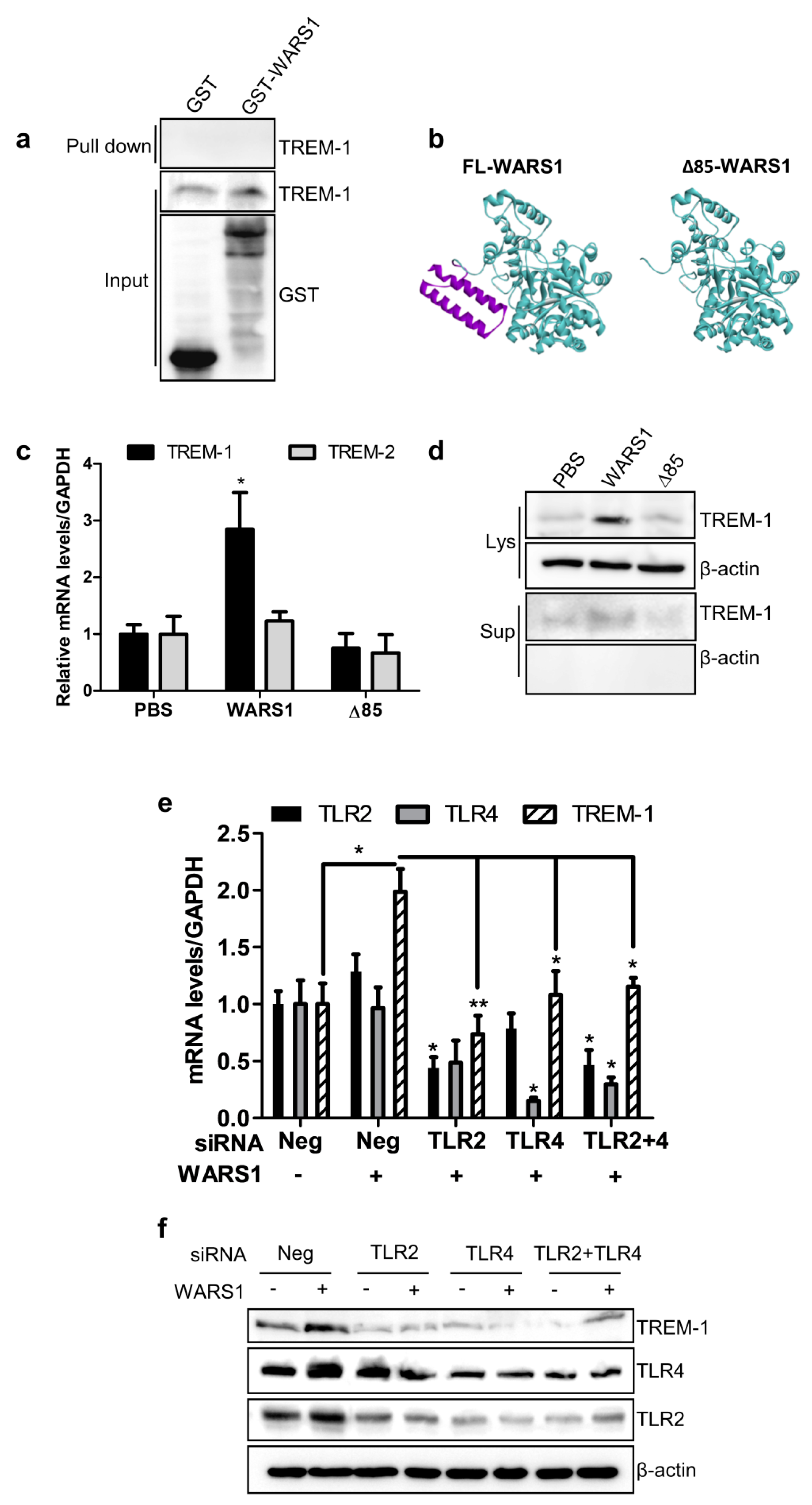
3.3. WARS1 Activated TREM-1 Expression via TLR4 and TLR2
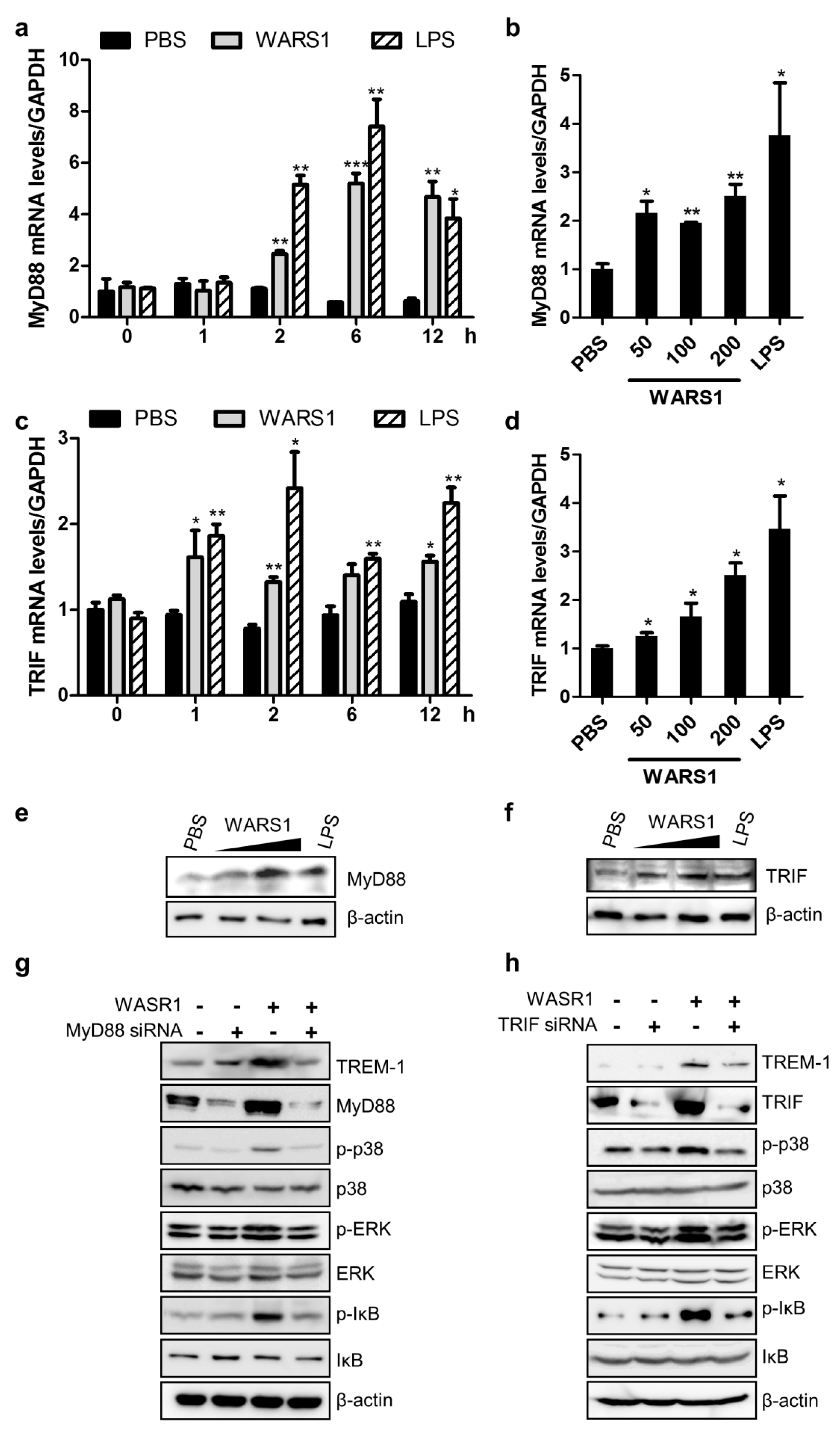
3.4. WARS1 Facilitated Both TRIF and MyD88 for TREM-1 Expression
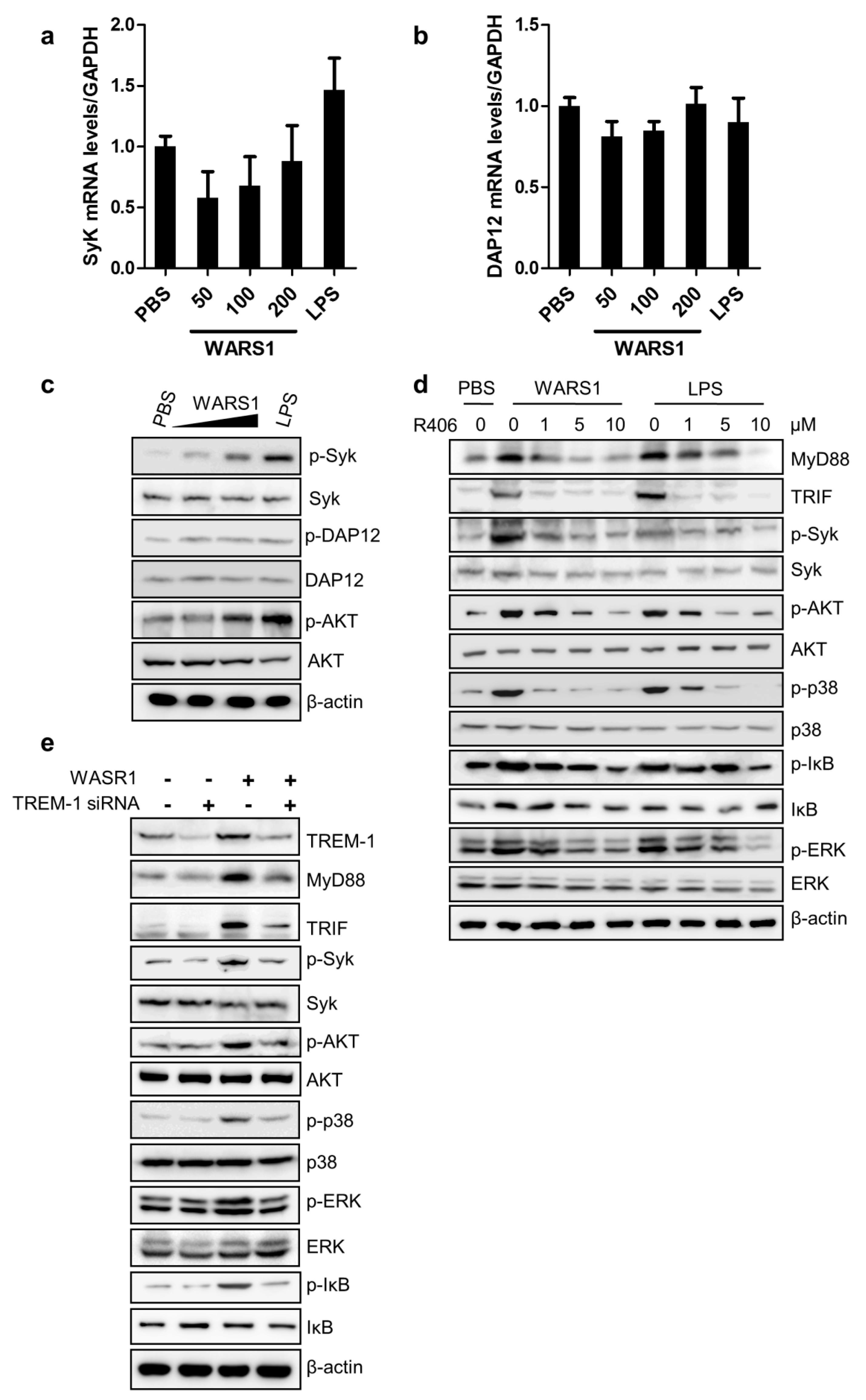
3.5. WARS1 Activated TREM-1 Signaling
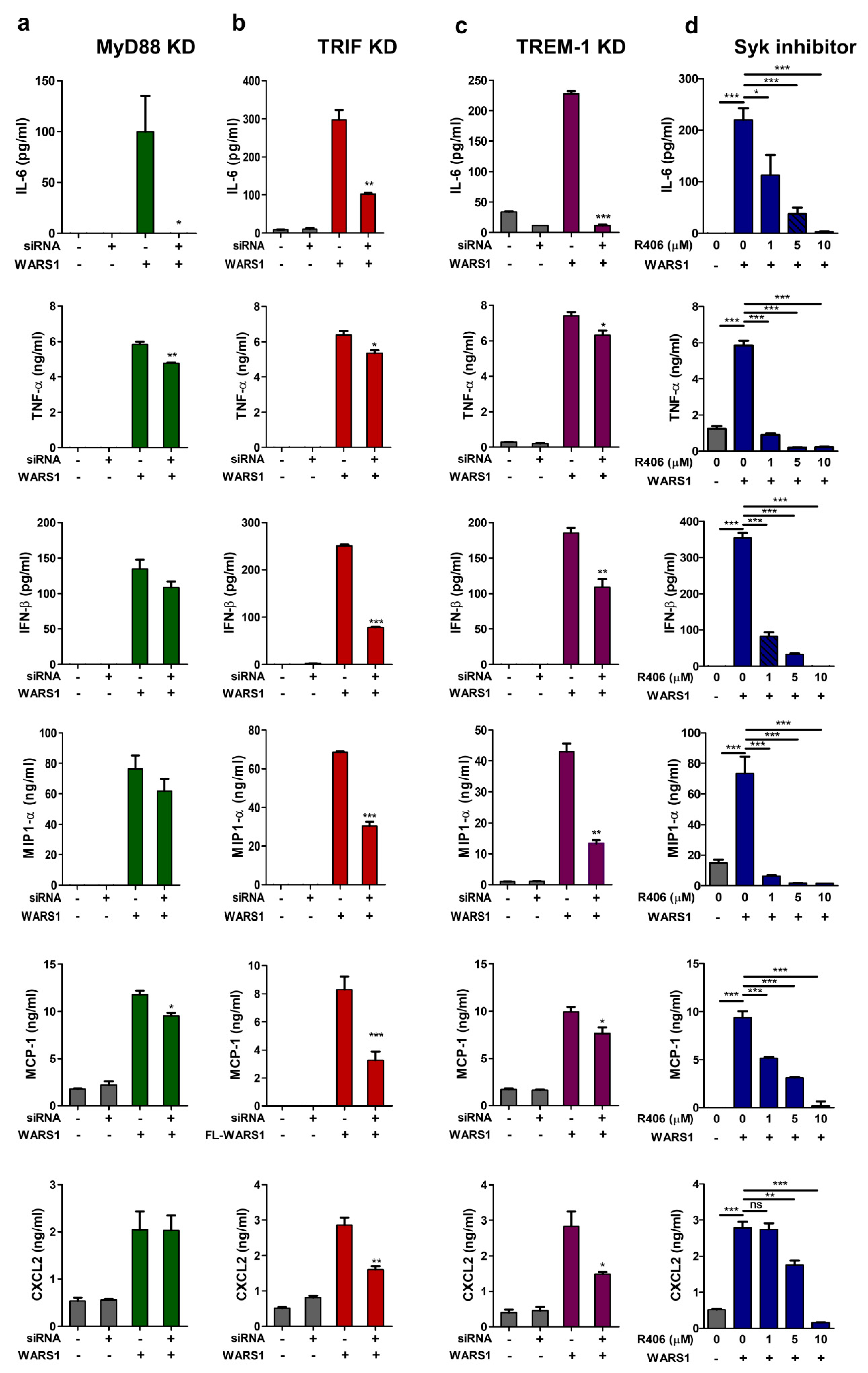
3.6. WARS1 Promoted Cytokine and Chemokine Production by the Cooperation of MyD88-TRIF-TREM-1 Signaling
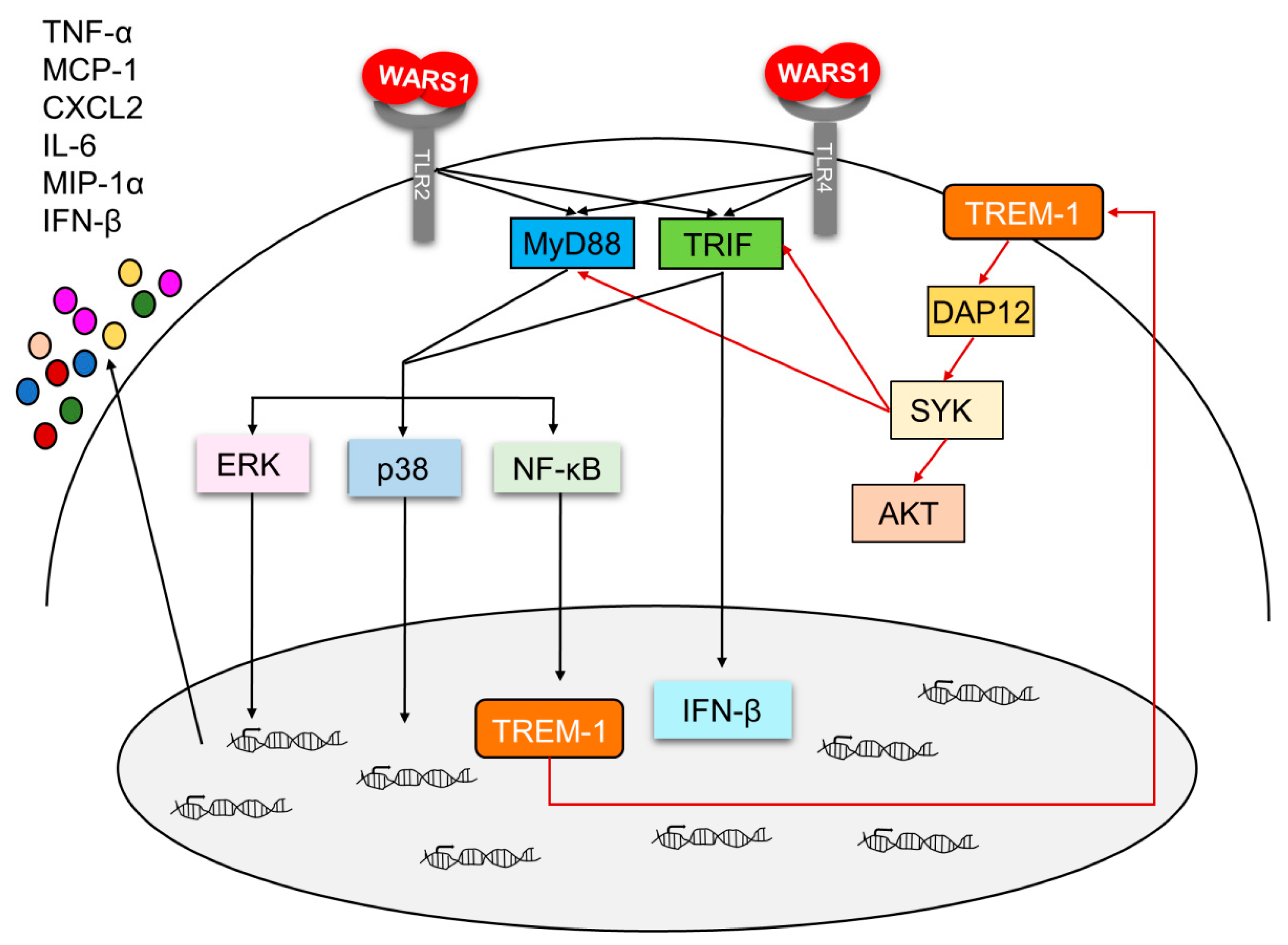
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kisselev, L.L. Mammalian tryptophanyl-tRNA synthetases. Biochimie 1993, 75, 1027–1039. [Google Scholar] [CrossRef]
- Kwon, N.H.; Fox, P.L.; Kim, S. Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discov. 2019, 18, 629–650. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Park, S.; Choi, J.J.; Park, B.K.; Rhee, K.H.; Kang, E.; Ahn, S.; Lee, C.-H.; Lee, J.S.; Inn, K.-S.; et al. Secreted tryptophanyl-tRNA synthetase as a primary defence system against infection. Nat. Microbiol. 2016, 2, 16191. [Google Scholar]
- Jin, M. Unique roles of tryptophanyl-tRNA synthetase in immune control and its therapeutic implications. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Lee, E.S.; Uddin, M.B.; Kim, T.H. Tryptophanyl-tRNA synthetase stimulates innate immune responses against viral infection. J. Virol. 2019, 93, e01291-e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, K.; Gibot, S.; Verma, S. Triggering receptor expressed on myeloid cells-1 (TREM-1): A new player in antiviral immunity? Front. Microbiol. 2014, 5, 627. [Google Scholar] [CrossRef] [Green Version]
- Pelham, C.J.; Pandya, A.N.; Agrawal, D.K. Triggering receptor expressed on myeloid cells receptor family modulators: A patent review. Expert Opin. Ther. Pat. 2014, 24, 1383–1395. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, S.J.; Simpson, T.; Schnapp, L.M. Triggering receptor expressed on myeloid cells type 1 as a potential therapeutic target in sepsis. Dimens. Crit. Care Nurs. 2012, 31, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2007, 14, 12. [Google Scholar] [CrossRef]
- Zheng, H.; Heiderscheidt, C.A.; Joo, M. MyD88-dependent and -independent activation of TREM-1 via specific TLR ligands. Eur. J. Immunol. 2010, 40, 162–171. [Google Scholar] [CrossRef]
- Carrasco, K.; Boufenzer, A.; Jolly, L.L.; Cordier, H. TREM-1 multimerization is essential for its activation on monocytes and neutrophils. Cell Mol. Immunol. 2019, 16, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, A.; Derive, M.; Gibot, S.; Leemans, J.C.; Florquin, S.; Dessing, M.C. TREM-1 and its potential ligands in non-infectious diseases: From biology to clinical perspectives. Pharmacol. Ther. 2017, 177, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Son, K.; Arauz, E.; Han, J.M.; Kim, S.H.; Chen, J. Leucyl-tRNA synthetase activates Vps34 in amino acid sensing. Cell Rep. 2016, 16, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.R.; Kim, S.R.; Im, J.B.; Lim, S.; Hong, I.S. Tryptophanyl-tRNA synthetase, a novel damage-induced cytokine, significantly increases the therapeutic effects of endometrial stem cells. Cytotherapy 2020, 22, 98. [Google Scholar] [CrossRef]
- Dubar, M.; Carrasco, K.; Gibot, S.; Bisson, C. Effects of Porphyromonasgingivalis LPS and LR12 peptide on TREM-1 expression by monocytes. J. Clin. Period Ontol. 2018, 45, 799–805. [Google Scholar]
- Klesney-Tait, J.; Turnbullm, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273. [Google Scholar] [CrossRef]
- Arts, R.J.; Joosten, L.A.; van der Meer, J.W. TREM-1: Intracellular signaling pathways and interaction with pattern recognition receptors. J. Leukoc. Biol. 2013, 93, 209–215. [Google Scholar] [CrossRef]
- Bleharski, J.R.; Kiessler, V.; Buonsanti, C. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J. Immunol. 2003, 170, 3812–3818. [Google Scholar] [CrossRef]
- Jennifer, R.T.; Marcus, J.K.; Cameron, P.S. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar]
- Shin, J.; Jin, M. Potential immunotherapeutics for immunosuppression in sepsis. Biomol. Ther. (Seoul) 2017, 25, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, H.; Zhou, J.; Xhong, Y. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar] [PubMed]
- Choi, J.S.; Yoon, B.R.; Shin, J.H.; Lee, S.H. Clinical value of full-length tryptophanyl-tRNA synthetase for sepsis detection in critically ill patients—A retrospective clinical assessment. Int. J. Infect. Dis. 2020, 97, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Kim, S.; Kim, M.H. Aminoacyl-tRNA synthetases, therapeutic targets for infectious diseases. Biochem. Pharmacol. 2018, 154, 424–434. [Google Scholar] [CrossRef]
- Joffre, J.; Potteaux, S.; Zeboudj, L. Genetic and pharmacological inhibition of TREM-1 limits the development of experimental atherosclerosis. J. Am. Coll. Cardiol. 2016, 68, 2776–2793. [Google Scholar] [CrossRef] [PubMed]
- Radsak, M.P.; Salih, H.R.; Rammensee, H.G. Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: Differential regulation of activation and survival. J. Immunol. 2014, 172, 4956–4963. [Google Scholar] [CrossRef] [Green Version]
- Boufenzer, A.; Lemarie, J.; Simon, T.; Derive, M. TREM-1 mediates inflammatory injury and cardiac remodeling following myocardial infarction. Circ. Res. 2015, 116, 1772–1782. [Google Scholar] [CrossRef] [Green Version]
- Cuvier, V.; Lorch, U.; Witte, S.; Olivier, A. A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition. J. Clin. Pharmacol. 2018, 84, 2270–2279. [Google Scholar] [CrossRef]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and crosstalk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef]
- Hammaker, D.; Firestein, G.S. “Go upstream, young man”: Lessons from the p38 saga. Ann. Rheum Dis. 2010, 69, i77–i82. [Google Scholar] [CrossRef]
- Tan, R.S.; Ho, B.; Leung, B.P.; Ding, J.L. TLR cross-talk confers specificity to innate immunity. Int. Rev. Immunol. 2014, 33, 443–453. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.T.; Yoon, H.K.; Kim, Y.T.; Choi, Y.H.; Lee, W.-K.; Jin, M. Tryptophanyl-tRNA Synthetase 1 Signals Activate TREM-1 via TLR2 and TLR4. Biomolecules 2020, 10, 1283. https://doi.org/10.3390/biom10091283
Nguyen TTT, Yoon HK, Kim YT, Choi YH, Lee W-K, Jin M. Tryptophanyl-tRNA Synthetase 1 Signals Activate TREM-1 via TLR2 and TLR4. Biomolecules. 2020; 10(9):1283. https://doi.org/10.3390/biom10091283
Chicago/Turabian StyleNguyen, Tram T. T., Hee Kyeong Yoon, Yoon Tae Kim, Yun Hui Choi, Won-Kyu Lee, and Mirim Jin. 2020. "Tryptophanyl-tRNA Synthetase 1 Signals Activate TREM-1 via TLR2 and TLR4" Biomolecules 10, no. 9: 1283. https://doi.org/10.3390/biom10091283
APA StyleNguyen, T. T. T., Yoon, H. K., Kim, Y. T., Choi, Y. H., Lee, W.-K., & Jin, M. (2020). Tryptophanyl-tRNA Synthetase 1 Signals Activate TREM-1 via TLR2 and TLR4. Biomolecules, 10(9), 1283. https://doi.org/10.3390/biom10091283