Activated Protein Kinase C (PKC) Is Persistently Trafficked with Epidermal Growth Factor (EGF) Receptor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Signaling from the EGFR
1.2. Does PKC Regulate EGFR Traffic?
2. Materials and Methods
2.1. Cell Culture and Nanoparticle Exposure
2.2. Preparation of Nanogold
2.3. Localization by Indirect Immunofluorescence
2.4. Image Acquisition and Analysis Procedures
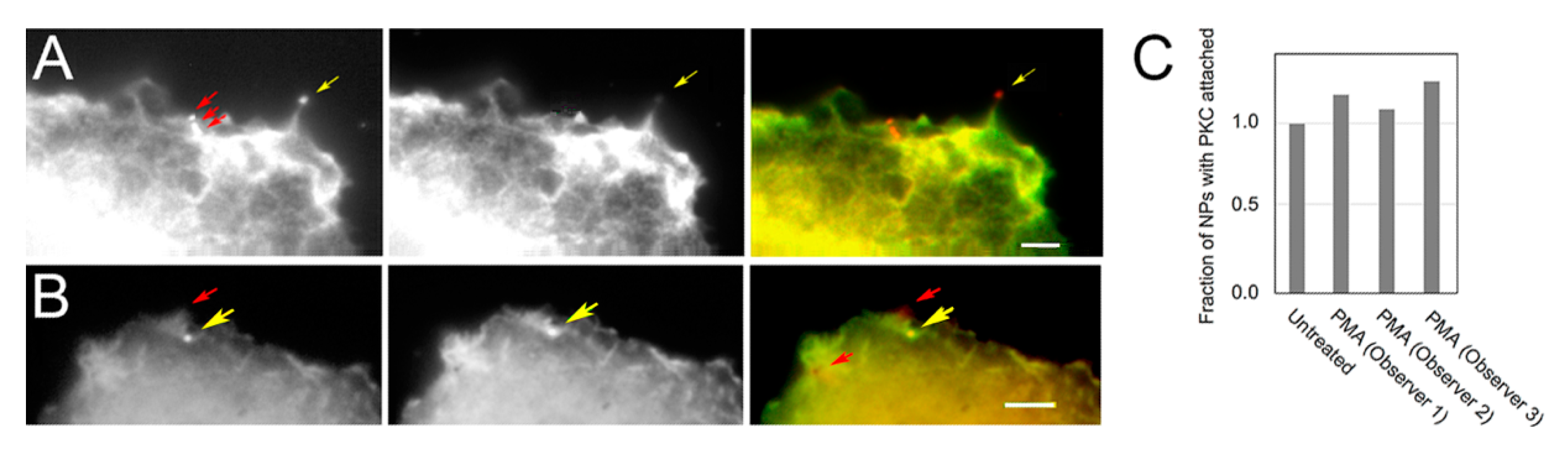
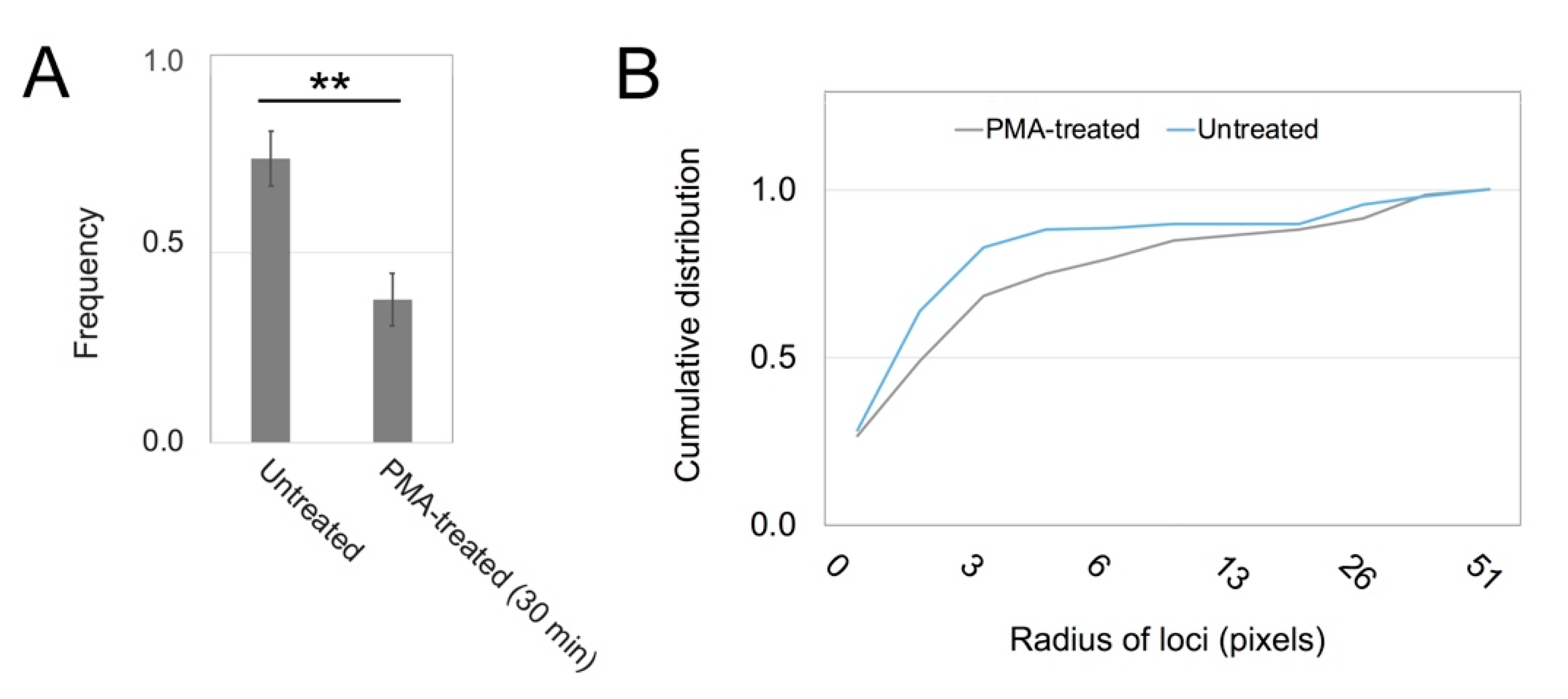
2.5. Image Quantification, Correlation Analysis, and Statistics
3. Results
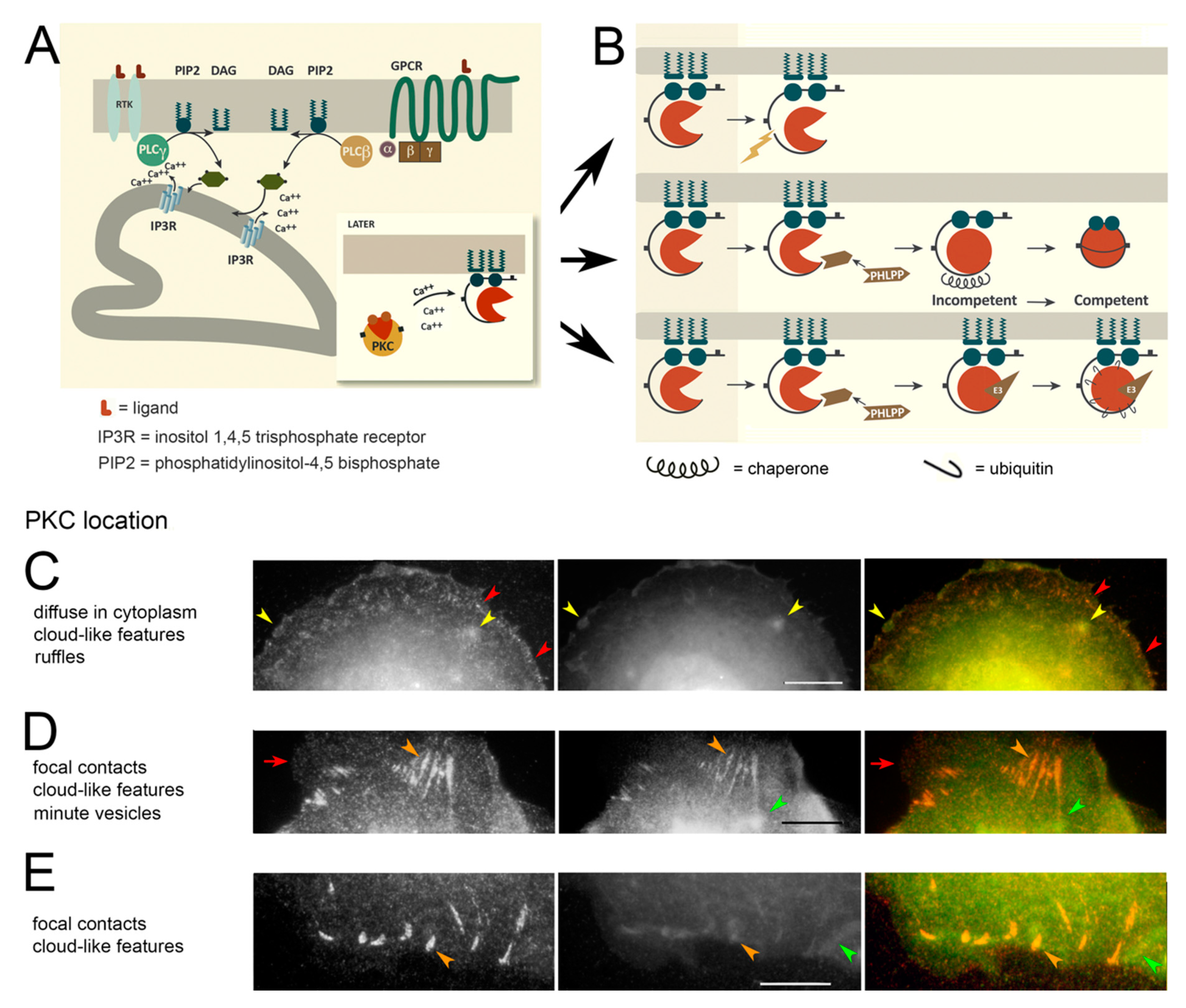
3.1. PKC Locations Are Dictated in Part by Actin Binding
3.2. PKC Is Recruited to Membranes with EGF-Tagged Gold Particles
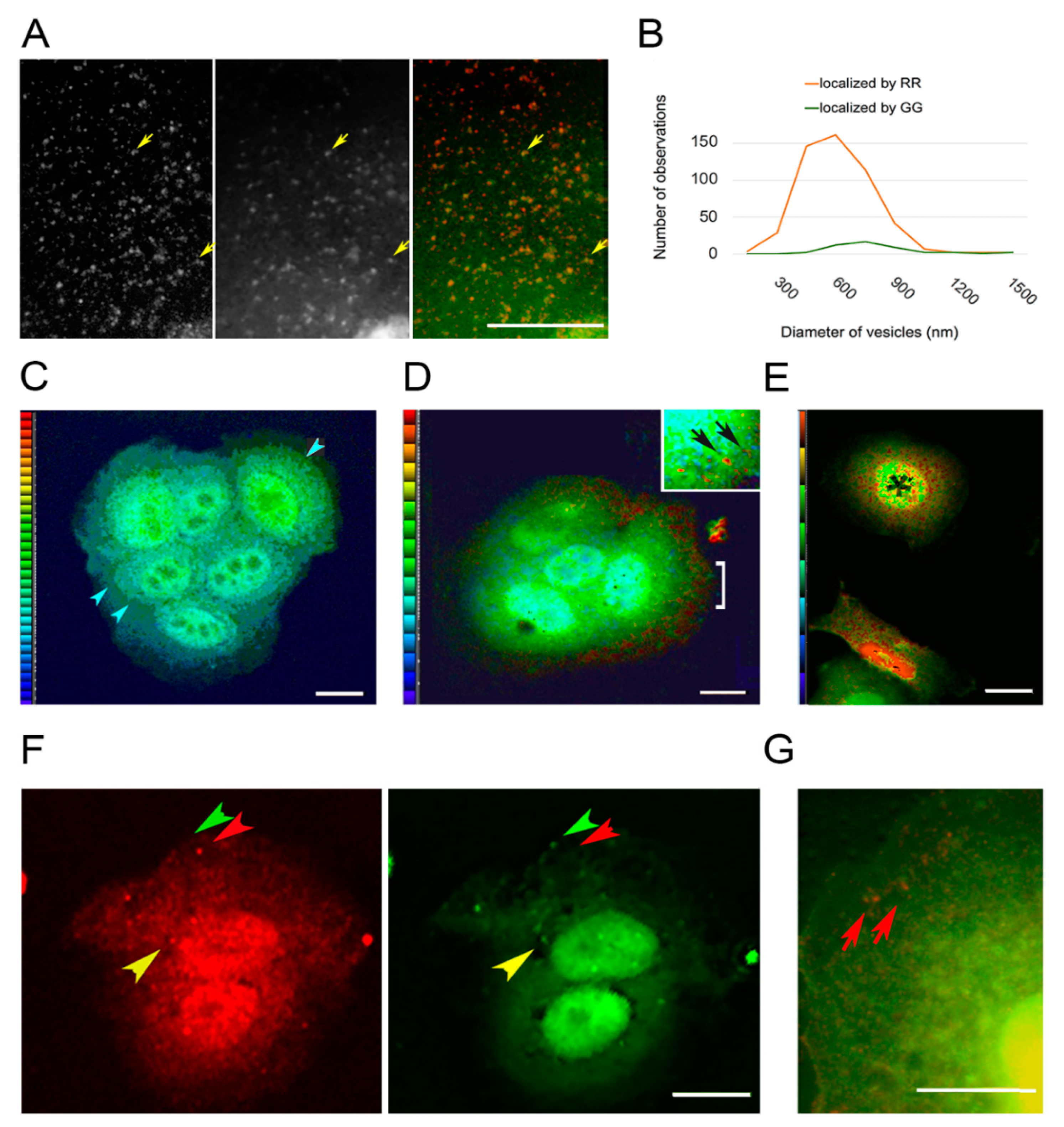
3.3. Vesicles Recruit PKC Displaying the RR Epitope and Retain It during Internalization
3.4. PKC Conversion from the Form Displaying the RR Epitope to the GG Form of the Molecule
4. Discussion
4.1. What Is the ERC?
4.2. PKC Localization
4.3. Relevance to Growth Control and Cancer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—Impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef]
- Van Der Steen, N.; Giovannetti, E.; Carbone, D.; Leonetti, A.; Rolfo, C.D.; Peters, G.J. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resist. 2018, 1, 230–249. [Google Scholar] [CrossRef]
- Heckman, C.A.; Wade, J.G. Protein kinase C: Its role in RTK processing. Trends Cancer Res. 2018, 13, 1–28. [Google Scholar]
- Lund, K.A.; Lazar, C.S.; Chen, W.S.; Walsh, B.J.; Welsh, J.B.; Herbst, J.J.; Walton, G.M.; Rosenfeld, M.G.; Gill, G.N.; Wiley, H.S. Phosphorylation of the epidermal growth factor receptor at threonine 654 inhibits ligand-induced internalization and down-regulation. J. Biol. Chem. 1990, 265, 20517–20523. [Google Scholar]
- Decker, S.J.; Ellis, C.; Pawson, T.; Velu, T. Effects of substitution of threonine 654 of the epidermal growth factor receptor on epidermal growth factor-mediated activation of phospholipase C. J. Biol. Chem. 1990, 265, 7009–7015. [Google Scholar]
- Seedorf, K.; Shearman, M.; Ullrich, A. Rapid and long term effects of protein kinase C on receptor tyrosine kinase phosphorylation and degradation. J. Biol. Chem. 1995, 270, 18953–18960. [Google Scholar] [CrossRef]
- Kluba, M.; Engelborghs, Y.; Hofkens, J.; Mizuno, H. Inhibition of receptor dimerization as a novel negative feedback mechanism of EGFR signaling. PLoS ONE 2015, 10, e0139971. [Google Scholar] [CrossRef]
- Liu, M.; Idkowiak-Baldys, J.; Roddy, P.L.; Baldys, A.; Raymond, J.; Clarke, C.J.; Hannun, Y.A. Sustained activation of protein kinase C induces delayed phosphorylation and regulates the fate of epidermal growth factor receptor. PLoS ONE 2013, 8, e80721. [Google Scholar] [CrossRef]
- Becker, K.P.; Hannun, Y.A. Isoenzyme-specific translocation of protein kinase C (PKC)βII and not PKCβI to a juxtanuclear subset of recycling endosomes: Involvement of phospholipase D. J. Biol. Chem. 2004, 279, 28251–28256. [Google Scholar] [CrossRef]
- Bao, J.; Alroy, I.; Waterman, H.; Schejter, E.D.; Brodie, C.; Gruenberg, J.; Yarden, Y. Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J. Biol. Chem. 2000, 275, 26178–26186. [Google Scholar] [CrossRef]
- Dietrich, M.; Malik, M.S.; Skeie, M.; Bertelsen, V.; Stang, E. Protein kinase C regulates ErbB3 turnover. Exp. Cell. Res. 2019, 382, 111473. [Google Scholar] [CrossRef]
- Bailey, T.A.; Luan, H.; Tom, E.; Bielecki, T.A.; Mohapatra, B.; Ahmad, G.; George, M.; Kelly, D.L.; Natarajan, A.; Raja, S.M.; et al. A kinase inhibitor screen reveals protein kinase C-dependent endocytic recycling of ErbB2 in breast cancer cells. J. Biol. Chem. 2014, 289, 30443–30458. [Google Scholar] [CrossRef]
- Raghunath, A.; Ling, M.; Larsson, C. The catalytic domain limits the translocation of protein kinase C alpha in response to increases in Ca2+ and diacylglycerol. Biochem. J. 2003, 370, 901–912. [Google Scholar] [CrossRef]
- Pu, Y.; Garfield, S.H.; Kedei, N.; Blumberg, P.M. Characterization of the differential roles of the twin C1a and C1b domains of protein kinase Cδ. J. Biol. Chem. 2009, 284, 1302–1312. [Google Scholar] [CrossRef]
- Heckman, C.A.; Runyeon, C.S.; Seubert, S.; Wade, J.G. Mathematical modeling of marker influx and efflux in cells. Bull Math Biol. 2001, 63, 431–439. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef]
- Idkowiak-Baldys, J.; Becker, K.P.; Kitatani, K.; Hannun, Y.A. Dynamic sequestration of the recycling compartment by classical protein kinase C. J. Biol. Chem. 2006, 281, 22321–22331. [Google Scholar] [CrossRef]
- Yamamoto, K.; Seki, T.; Yamamoto, H.; Adachi, N.; Tanaka, S.; Hide, I.; Saito, N.; Sakai, N. Deregulation of the actin cytoskeleton and macropinocytosis in response to phorbol ester by the mutant protein kinase C gamma that causes spinocerebellar ataxia type 14. Front. Physiol. 2014, 5, 126. [Google Scholar] [CrossRef]
- Schmid, S.L.; Sorkin, A.; Zerial, M. (Eds.) Endocytosis; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2014; 590p. [Google Scholar]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Naslavsky, N.; Caplan, S. The enigmatic endosome–sorting the ins and outs of endocytic trafficking. J. Cell Sci. 2016, 131, jcs216499. [Google Scholar] [CrossRef]
- Li, Y.; Urban, J.M.; Cayer, M.L.; Plummer, H.K., III; Heckman, C.A. Actin-based features negatively regulated by protein kinase C-epsilon. Am. J. Physiol. Cell Physiol. 2006, 291, C1002–C1013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klumperman, J.; Raposo, G. The complex ultrastructure of the endolysosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a016857. [Google Scholar] [CrossRef] [PubMed]
- Alvi, F.; Idkowiak-Baldys, J.; Baldys, A.; Raymond, J.R.; Hannun, Y.A. Regulation of membrane trafficking and endocytosis by protein kinase C: Emerging role of the pericentrion, a novel protein kinase C-dependent subset of recycling endosomes. Cell. Mol. Life Sci. 2007, 64, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef]
- Sakai, R.; Fukuda, R.; Unida, S.; Aki, M.; Ono, Y.; Endo, A.; Kusumi, S.; Koga, D.; Fukushima, T.; Komada, M.; et al. The integral function of the endocytic recycling compartment is regulated by RFFL-mediated ubiquitylation of Rab11 effectors. J. Cell Sci. 2019, 132, jcs228007. [Google Scholar] [CrossRef]
- Marchok, A.C.; Rhoton, J.C.; Griesemer, R.A.; Nettesheim, P. Increased in vitro growth capacity of tracheal epithelium exposed in vivo to 7,12-dimethylbenz(a)anthracene. Cancer Res. 1977, 37, 1811–1821. [Google Scholar]
- Heckman, C.A.; Varghese, M.; Cayer, M.L.; Boudreau, N.S. Origin of ruffles: Linkage to other protrusions, filopodia and lamellae. Cell. Signal. 2012, 24, 189–198. [Google Scholar] [CrossRef]
- Heckman, C.A.; Pandey, P.; Cayer, M.L.; Biswas, T.; Zhang, Z.-Y.; Boudreau, N.S. The tumor promoter-activated protein kinase Cs are a system for regulating filopodia. Cytoskeleton 2017, 74, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bao, L.; Hyun, B.-R.; Bartnik, A.C.; Zhong, Y.-W.; Reed, J.C.; Pang, D.-W.; Abruña, H.D.; Malliaras, G.G.; Wise, F.W. Electrogenerated chemiluminescence from PbS quantum dots. Nano Lett. 2009, 9, 789–793. [Google Scholar] [CrossRef]
- Newton, A.C. Regulation of conventional and novel protein kinase C isozymes by phosphorylation and lipids. In Protein Kinase C in Cancer Signaling and Therapy; Kazanietz, M.G., Ed.; Springer Science: New York, NY, USA, 2010; pp. 9–23. [Google Scholar]
- Swanson, C.J.; Ritt, M.; Wang, W.; Lang, M.J.; Narayan, A.; Tesmer, J.J.; Westfall, M.; Sivaramakrishnan, S. Conserved modular domains team up to latch-open active protein kinase Cα. J. Biol. Chem. 2014, 289, 17812–17829. [Google Scholar] [CrossRef] [PubMed]
- Leonard, T.A.; Różycki, B.; Saidi, L.F.; Hummer, G.; Hurley, J.H. Crystal structure and allosteric activation of protein kinase C βII. Cell 2011, 144, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, D.; Braiman-Wiksman, L.; Daum, N.; Tennenbaum, T. The Protein Kinase C Family: Key Regulators Bridging Signaling Pathways in Skin and Tumor Epithelia. In Post-Translational Modifications in Health and Disease; Vidal, C., Ed.; Springer: New York, NY, USA, 2011; Volume 13, pp. 171–198. [Google Scholar]
- Laummonerie, C.; Mutterer, J. Colocalization Finder (ImageJ Plugin). 2004. Available online: http://rsbweb.nih.gov/ij/plugins/colocalization-finder.html (accessed on 21 July 2020).
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Weekes, J.; Barry, S.T.; Critchley, D.R. Acidic phospholipids inhibit the intramolecular association between the N- and C-terminal regions of vinculin, exposing actin-binding and protein kinase C phosphorylation sites. Biochem. J. 1996, 314, 827–832. [Google Scholar] [CrossRef]
- Thievessen, I.; hompson, P.M.; Berlemont, S.; Plevock, K.M.; Plotnikov, S.V.; Zemljic-Harpf, A.; Ross, R.S.; Davidson, M.W.; Danuser, G.; Campbell, S.L.; et al. Vinculin–actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. J. Cell Biol. 2013, 202, 163–177. [Google Scholar] [CrossRef]
- Lidke, D.S.; Nagy, P.; Heintzmann, R.; Arndt-Jovin, D.J.; Post, J.N.; Grecco, H.E.; Jares-Erijman, E.A.; Jovin, T.M. Quantum dot ligands provide new insights into erbB/HER receptor-mediated signal transduction. Nat. Biotechnol. 2004, 22, 198–203. [Google Scholar] [CrossRef]
- Lidke, D.S.; Lidke, K.A.; Rieger, B.; Jovin, T.M.; Arndt-Jovin, D.J. Reaching out for signals: Filopodia sense EGF and respond by directed retrograde transport of activated receptors. J. Cell Biol. 2005, 170, 619–626. [Google Scholar] [CrossRef]
- Sharma, G.; Pallesen, J.; Das, S.; Grassucci, R.; Langlois, R.; Hampton, C.M.; Kelly, D.F.; des Georges, A.; Frank, J. Affinity grid-based cryo-EM of PKC binding to RACK1 on the ribosome. J. Struct. Biol. 2013, 181, 190–194. [Google Scholar] [CrossRef]
- Lin, S.X.; Mallet, W.G.; Huang, A.Y.; Maxfield, F.R. Endocytosed cation-Independent mannose 6-phosphate receptor traffics via the endocytic recycling compartment en route to the trans-Golgi network and a subpopulation of late endosomes. Mol. Biol. Cell 2004, 15, 721–733. [Google Scholar] [CrossRef]
- Xie, S.; Bahl, K.; Reinecke, J.S.; Hammond, G.R.V.; Naslavsky, N.; Caplana, S. The endocytic recycling compartment maintains cargo segregation acquired upon exit from the sorting endosome. Mol. Biol. Cell 2016, 27, 108–126. [Google Scholar] [CrossRef]
- Lin, S.X.; Gundersen, G.G.; Maxfield, F.R. Export from pericentriolar endocytic recycling compartment to cell surface depends on stable, detyrosinated (Glu) microtubules and kinesin. Mol. Biol. Cell 2002, 13, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Mahmutefendić, H.; Zagorac, G.B.; Maćešić, S.; Lučin, P. Rapid Endosomal Recycling. In Peripheral Membrane Proteins; Tanabe, S., Ed.; IntechOpen Ltd.: London, UK, 2018. [Google Scholar] [CrossRef]
- Das, S.; Pellett, P.E. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 2011, 85, 5864–5879. [Google Scholar] [CrossRef] [PubMed]
- Lučin, P.; Kareluša, L.; Zagorac, G.B.; Lučin, H.M.; Pavišić, V.; Vučko, N.J.; Lukanović Jurić, S.L.; Marcelić, M.; Lisnić, B.; Jonjić, S. Cytomegaloviruses exploit recycling Rab proteins in the sequential establishment of the assembly compartment. Front. Cell Dev. Biol. 2018, 6, 165. [Google Scholar] [CrossRef]
- Taisne, C.; Lussignol, M.; Hernandez, E.; Moris, A.; Mouna, L.; Esclatine, A. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci. Rep. 2019, 9, 4560. [Google Scholar] [CrossRef] [PubMed]
- Kaleem, A.; Ahmad, I.; Hoessli, D.C.; Walker-Nasir, E.; Saleem, M.; Shakoori, A.R.; Din, N. Epidermal growth factor receptors: Function modulation by phosphorylation and glycosylation interplay. Mol. Biol. Rep. 2009, 36, 631–639. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taunton, J.; Rowning, B.A.; Coughlin, M.L.; Wu, M.; Moon, R.T.; Mitchison, T.J.; Larabell, C.A. Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J. Cell Biol. 2000, 148, 519–530. [Google Scholar] [CrossRef]
- Lladó, A.; Timpson, P.; Moretó, J.; Pol, A.; Grewal, T.; Daly, R.J.; Enrich, C.; Tebar, F. Protein kinase Cδ and calmodulin regulate epidermal growth factor receptor recycling from early endosomes through Arp2/3 complex and cortactin. Mol. Biol. Cell 2008, 19, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Koese, M.; Rentero, C.; Kota, B.P.; Hoque, M.; Cairns, R.; Wood, P.; Vilà de Muga, S.; Reverter, M.; Alvarez-Guaita, A.; Monastyrskaya, K.; et al. Annexin A6 is a scaffold for PKCα to promote EGFR inactivation. Oncogene 2013, 32, 2858–2872. [Google Scholar] [CrossRef]
- Gould, C.M.; Newton, A.C. The life and death of protein kinase C. Curr. Drug Targets 2008, 9, 614–625. [Google Scholar] [CrossRef]
- Gao, T.; Brognard, J.; Newton, A.C. The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 2008, 283, 6300–6311. [Google Scholar] [CrossRef]
- De Melker, A.A.; van der Horst, G.; Calafat, J.; Jansen, H.; Borst, J. c-Cbl ubiquitinates the EGF receptor at the plasma membrane and remains receptor associated throughout the endocytic route. J. Cell Sci. 2001, 114, 2167–2178. [Google Scholar] [PubMed]
- Wolf-Yadlin, A.; Hautaniemi, S.; Lauffenburger, D.A.; White, F.M. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. USA 2007, 104, 5860–5865. [Google Scholar] [CrossRef] [PubMed]
- Heibeck, T.H.; Ding, S.-J.; Opresko, L.K.; Zhao, R.; Schepmoes, A.A.; Yang, F.; Tolmachev, A.V.; Monroe, M.E.; Camp, D.G., II; Smith, R.D.; et al. An extensive survey of tyrosine phosphorylation revealing new sites in human mammary epithelial cells. J. Proteome Res. 2009, 8, 3852–3861. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olsen, V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Wandinger, S.K.; Lahortiga, I.; Jacobs, K.; Klammer, M.; Jordan, N.; Elschenbroich, S.; Parade, M.; Jacoby, E.; Linders, J.T.M.; Brehmer, D.; et al. Quantitative phosphoproteomics analysis of ERBB3/ERBB4 signaling. PLoS ONE 2016, 11, e0146100. [Google Scholar] [CrossRef]
- Kedei, N.; Lundberg, D.J.; Toth, A.; Welburn, P.; Garfield, S.H.; Blumberg, P.M. Characterization of the interaction of ingenol 3-angelate with protein kinase C. Cancer Res. 2004, 64, 3243–3255. [Google Scholar] [CrossRef]
- Sakai, N.; Sasaki, K.; Ikegaki, N.; Shirai, Y.; Ono, Y.; Saito, N. Direct visualization of the translocation of the gamma-subspecies of protein kinase C in living cells using fusion proteins with green fluorescent protein. J. Cell Biol. 1997, 139, 1465–1476. [Google Scholar] [CrossRef]
- Brown, S.G.; Thomas, A.; Dekker, L.V.; Tinker, A.; Leaney, J.L. PKC-δ sensitizes Kir3.1/3.2 channels to changes in membrane phospholipid levels after M3 receptor activation in HEK-293 cells. Am. J. Physiol. Cell Physiol. 2005, 289, C543–C556. [Google Scholar] [CrossRef]
- Goedhart, J.; Gadella, T.W.J. Fluorescence resonance energy transfer imaging of PKC signalling in living cells using genetically encoded fluorescent probes. J. Neurosci. 2009, 30. [Google Scholar] [CrossRef]
- Collazos, A.; Diouf, B.; Guérineau, N.C.; Quittau-Prévostel, C.; Peter, M.; Coudane, F.; Hollande, F.; Joubert, D. A spatiotemporally coordinated cascade of protein kinase C activation controls isoform-selective translocation. Mol. Cell. Biol. 2006, 26, 2247–2261. [Google Scholar] [CrossRef]
- Kang, J.-H. Protein kinase C (PKC) isozymes and cancer. New J. Sci. 2014, 231418, 36. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.; Sabo, E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids 2008, 73, 870–873. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heckman, C.A.; Biswas, T.; Dimick, D.M.; Cayer, M.L. Activated Protein Kinase C (PKC) Is Persistently Trafficked with Epidermal Growth Factor (EGF) Receptor. Biomolecules 2020, 10, 1288. https://doi.org/10.3390/biom10091288
Heckman CA, Biswas T, Dimick DM, Cayer ML. Activated Protein Kinase C (PKC) Is Persistently Trafficked with Epidermal Growth Factor (EGF) Receptor. Biomolecules. 2020; 10(9):1288. https://doi.org/10.3390/biom10091288
Chicago/Turabian StyleHeckman, Carol A., Tania Biswas, Douglas M. Dimick, and Marilyn L. Cayer. 2020. "Activated Protein Kinase C (PKC) Is Persistently Trafficked with Epidermal Growth Factor (EGF) Receptor" Biomolecules 10, no. 9: 1288. https://doi.org/10.3390/biom10091288
APA StyleHeckman, C. A., Biswas, T., Dimick, D. M., & Cayer, M. L. (2020). Activated Protein Kinase C (PKC) Is Persistently Trafficked with Epidermal Growth Factor (EGF) Receptor. Biomolecules, 10(9), 1288. https://doi.org/10.3390/biom10091288