Structural Models for the Dynamic Effects of Loss-of-Function Variants in the Human SIM1 Protein Transcriptional Activation Domain

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computer-Assisted Modeling of SIM1 and ARNT Protein Structures
2.2. Molecular Dynamics Simulations
3. Results
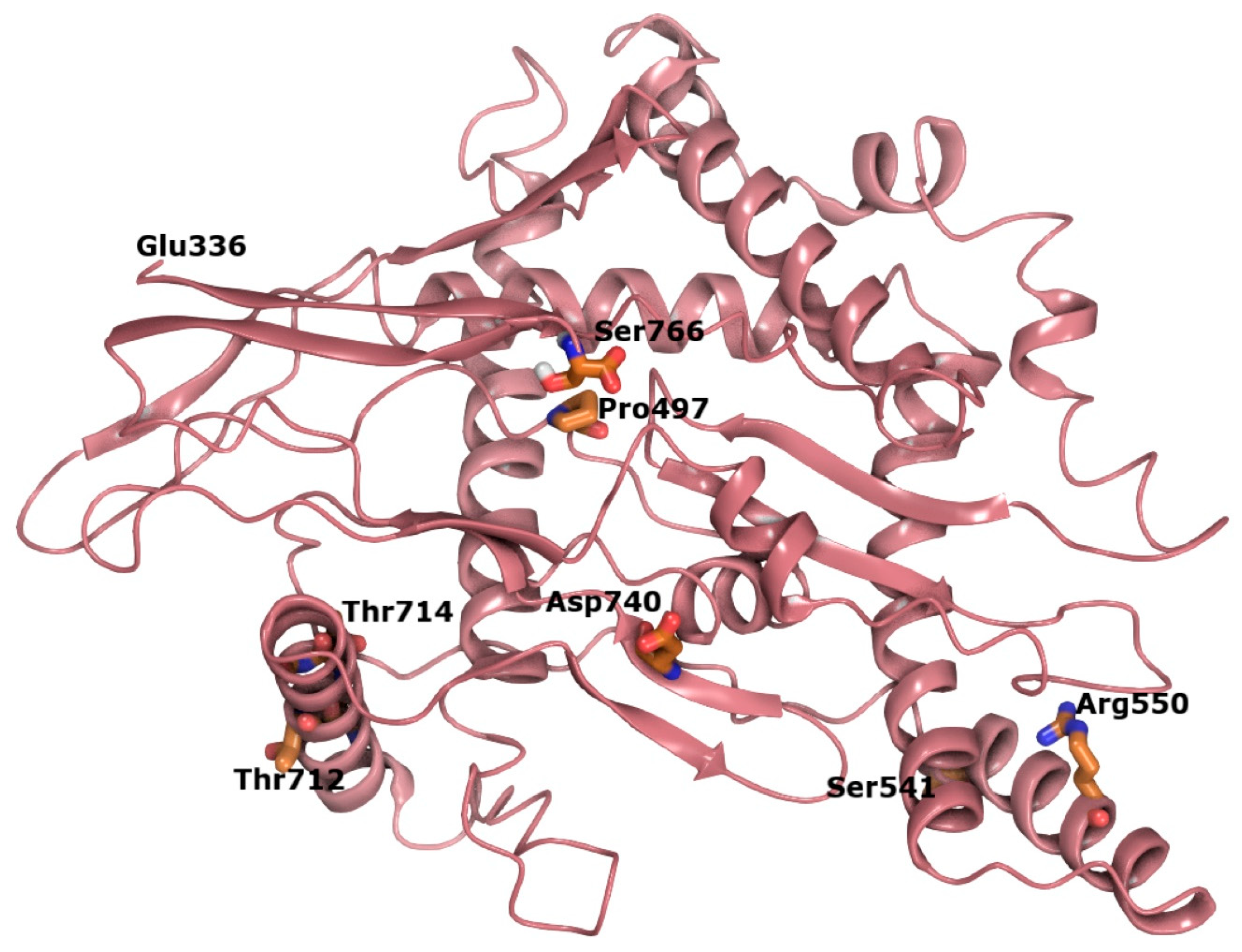
3.1. SIM1 and ARNT Heterodimer Interface Has Contacts Consistent with Stabilizing Interface
3.1.1. SIM1 bHLH Domain
3.1.2. SIM1 PASA Domain
3.1.3. SIM1 PASB Domain
3.1.4. SIM1 PAC Domain
3.1.5. SM Domain of SIM1 and ARNT
3.2. SIM1 Dynamics Demonstrates Pathogenic Variants Have Both Local and Global Effects
3.2.1. T46R Variant
3.2.2. D707H Variant
3.2.3. G715V Variant
3.2.4. D740H Variant
3.3. Detailed Analyses for Local Deviations in Geometry Lead to Larger Amplitude Changes via Correlated Motions
3.4. Particle Size Changes as a Consequence of the Variant Chosen
3.5. Stabilization of the Local Region Shifts through Hydrogen Bonding Network Disruptions (Triggering the Correlated Motion Cascade)
3.6. Apo SIM1 Has Room to Move Forming Intra-Molecular Interactions
4. Discussion
4.1. Mutant T46R
4.2. Mutant H707D
4.3. Mutant H740D
4.4. Mutant G715V
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Henry, J.T.; Crosson, S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 2011, 65, 261–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, J.L.; Rosenquist, T.; May, N.R.; Fan, C.M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998, 12, 3264–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, J.L.; Boucher, F.; Melnyk, A.; Gauthier, F.; Goshu, E.; Levy, E.; Mitchell, G.A.; Himms-Hagen, J.; Fan, C.M. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet. 2001, 10, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Xi, D.; Gandhi, N.; Lai, M.; Kublaoui, B.M. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PLoS ONE 2012, 7, e36453. [Google Scholar] [CrossRef]
- Elena, G.; Bruna, C.; Benedetta, M.; Stefania, D.C.; Giuseppe, C. Prader-willi syndrome: Clinical aspects. J. Obes. 2012, 2012, 473941. [Google Scholar] [CrossRef] [Green Version]
- Bonnefond, A.; Raimondo, A.; Stutzmann, F.; Ghoussaini, M.; Ramachandrappa, S.; Bersten, D.C.; Durand, E.; Vatin, V.; Balkau, B.; Lantieri, O.; et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J. Clin. Investig. 2013, 123, 3037–3041. [Google Scholar] [CrossRef] [Green Version]
- Woods, S.L.; Whitelaw, M.L. Differential activities of murine single minded 1 (SIM1) and SIM2 on a hypoxic response element. Cross-talk between basic helix-loop-helix/per-Arnt-Sim homology transcription factors. J. Biol. Chem. 2002, 277, 10236–10243. [Google Scholar] [CrossRef] [Green Version]
- Ramachandrappa, S.; Raimondo, A.; Cali, A.M.; Keogh, J.M.; Henning, E.; Saeed, S.; Thompson, A.; Garg, S.; Bochukova, E.G.; Brage, S.; et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J. Clin. Investig. 2013, 123, 3042–3050. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, P.R.; Sullivan, A.E.; Gerassimou, A.G.; Kleinendorst, L.; Bersten, D.C.; Cooiman, M.; Harris, K.G.; Wierenga, K.J.; Klee, E.W.; Van Gerpen, J.A.; et al. Functional Analysis of the SIM1 Variant p.G715V in 2 Patients With Obesity. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.; Sander, C.; Scharf, M.; Vriend, G. The PDBFINDER database: A summary of PDB, DSSP and HSSP information with added value. Comput. Appl. Biosci. 1996, 12, 525–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooft, R.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef] [PubMed]
- King, R.D.; Sternberg, M.J. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 1996, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Elber, R. SSALN: An alignment algorithm using structure-dependent substitution matrices and gap penalties learned from structurally aligned protein pairs. Proteins 2006, 62, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.A.; Keil, M.; Baker, D.S. Comparison of Composer and ORCHESTRAR. Proteins 2008, 72, 1243–1258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Pandit, S.B.; Skolnick, J. Performance of the Pro-sp3-TASSER server in CASP8. Proteins 2009, 77 (Suppl. 9), 123–127. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Skolnick, J. Protein structure prediction by pro-Sp3-TASSER. Biophys. J. 2009, 96, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Canutescu, A.A.; Shelenkov, A.A.; Dunbrack, R.L., Jr. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003, 12, 2001–2014. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef] [Green Version]
- Muckstein, U.; Hofacker, I.L.; Stadler, P.F. Stochastic pairwise alignments. Bioinformatics 2002, 18 (Suppl. 2), S153–S160. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Schrödinger. Biologics Suite, BioLuminate, Version 1.1; Schrödinger, LLC: New York, NY, USA, 2013. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38. [Google Scholar] [CrossRef]
- Caulfield, T.; Devkota, B. Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 2012. [Google Scholar] [CrossRef]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef]
- Caulfield, T.R. Inter-ring rotation of apolipoprotein A-I protein monomers for the double-belt model using biased molecular dynamics. J. Mol. Graph. Model. 2011, 29, 1006–1014. [Google Scholar] [CrossRef]
- Caulfield, T.R.; Devkota, B.; Rollins, G.C. Examinations of tRNA Range of Motion Using Simulations of Cryo-EM Microscopy and X-ray Data. J. Biophys. 2011, 2011, 219515. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Chem. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Reblova, K.; Lankas, F.; Razga, F.; Krasovska, M.V.; Koca, J.; Sponer, J. Structure, dynamics, and elasticity of free 16s rRNA helix 44 studied by molecular dynamics simulations. Biopolymers 2006, 82, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Reblova, K.; Fadrna, E.; Sarzynska, J.; Kulinski, T.; Kulhanek, P.; Ennifar, E.; Koca, J.; Sponer, J. Conformations of flanking bases in HIV-1 RNA DIS kissing complexes studied by molecular dynamics. Biophys. J. 2007, 93, 3932–3949. [Google Scholar] [CrossRef] [Green Version]
- Hines, S.L.; Mohammad, A.N.; Jackson, J.; Macklin, S.; Caulfield, T.R. Integrative data fusion for comprehensive assessment of a novel CHEK2 variant using combined genomics, imaging, and functional-structural assessments via protein informatics. Mol. Omics 2019, 15, 59–66. [Google Scholar] [CrossRef]
- Cheatham, T.E., III; Young, M.A. Molecular dynamics simulation of nucleic acids: Successes, limitations and promise. Biopolymers 2001, 56, 232–256. [Google Scholar] [CrossRef]
- Caulfield, T.R.; Fiesel, F.C.; Moussaud-Lamodiere, E.L.; Dourado, D.F.; Flores, S.C.; Springer, W. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput. Biol. 2014, 10, e1003935. [Google Scholar] [CrossRef] [Green Version]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2016. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Caulfield, T.; Xu, Y.F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013. [Google Scholar] [CrossRef]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coban, M.A.; Blackburn, P.R.; Whitelaw, M.L.; Haelst, M.M.v.; Atwal, P.S.; Caulfield, T.R. Structural Models for the Dynamic Effects of Loss-of-Function Variants in the Human SIM1 Protein Transcriptional Activation Domain. Biomolecules 2020, 10, 1314. https://doi.org/10.3390/biom10091314
Coban MA, Blackburn PR, Whitelaw ML, Haelst MMv, Atwal PS, Caulfield TR. Structural Models for the Dynamic Effects of Loss-of-Function Variants in the Human SIM1 Protein Transcriptional Activation Domain. Biomolecules. 2020; 10(9):1314. https://doi.org/10.3390/biom10091314
Chicago/Turabian StyleCoban, Mathew A., Patrick R. Blackburn, Murray L. Whitelaw, Mieke M. van Haelst, Paldeep S. Atwal, and Thomas R. Caulfield. 2020. "Structural Models for the Dynamic Effects of Loss-of-Function Variants in the Human SIM1 Protein Transcriptional Activation Domain" Biomolecules 10, no. 9: 1314. https://doi.org/10.3390/biom10091314
APA StyleCoban, M. A., Blackburn, P. R., Whitelaw, M. L., Haelst, M. M. v., Atwal, P. S., & Caulfield, T. R. (2020). Structural Models for the Dynamic Effects of Loss-of-Function Variants in the Human SIM1 Protein Transcriptional Activation Domain. Biomolecules, 10(9), 1314. https://doi.org/10.3390/biom10091314