1. Introduction
The single-minded homolog 1 (SIM1) is a founding member of a subset of basic helix-loop-helix (bHLH) proteins that also contain eriod (Per)-aryl hydrocarbon receptor nuclear transporter (ARNT)-SIM (PAS) domains. PAS domain proteins are universally expressed [
1], and they have an eclectic array of functions, such as chemotaxis, phototropism, ligand binding, and signal transduction [
1]. SIM1 is imperative for development and functionality of paraventricular neurons of the hypothalamus [
2], but it is also expressed in kidney and muscle, where its functions have yet to be investigated. As evidence for the importance of SIM1, homozygous null mice are perinatally lethal [
3]. Furthermore, SIM1 heterozygous mice develop juvenile obesity and associated diabetes-like symptoms via the etiological mechanism of metabolic imbalance through hyperphagia and lack of compensatory increase in energy output [
3,
4]. This phenotype is consistent with a crucial role for SIM1 in the leptin-melanocortin signaling pathway that promotes satiety via activation of paraventricular hypothalamic (PVH) neurons. SIM1 can form a heterodimer with either aryl hydrocarbon receptor nuclear translocator (ARNT) or ARNT2 in in vitro cell-based assays but is hypothesized to preferentially dimerize with ARNT2 in vivo. As ARNT2 is enriched in neurons, and
Arnt2−/− mice phenocopy the loss of PVH neurons observed in
Sim1−/− mice, it is hypothesized that disruption of the SIM1/ARNT2 axis drives obesity in humans with or without clinical features that are also described in Prader-Willi-like (PWL) syndrome. There are numerous pathogenic missense variants of
SIM1, though for many, the mechanism by which they exert dysfunction is unknown. A major contributing factor to this is the lack of a full-length, high-resolution experimental structure for SIM1, especially in complex with the aforementioned proteins.
Prader-Willi Syndrome (PWS) is a genetic disorder that is characterized by neonatal hypotonia and feeding problems, short stature, small hands and feet, impaired intellectual development, and from early childhood onwards, hyperphagia, which often results in obesity and diabetes. PWS is caused by lack of expression of genes on the paternally inherited chromosome 15q11.2-q13 region [
5]. Pathogenic
SIM1 variants often present with overlapping clinical features (hypotonia, hyperphagia, and variable degree of intellectual deficit), hereafter termed Prader-Willi-like (PWL). However, other
SIM1 variants are not associated with PWL features, but instead are only associated with early onset obesity with variable penetrance. It is currently unclear why certain
SIM1 variants result in PWL symptoms and others do not.
In this study, we sought to identify the mechanism by which several missense SIM1 variants effectuate pathological conditions through computational modeling, molecular dynamics simulations (MDS), and subsequent analyses. To that end, we generated an original atomistic model of SIM1 dimerized with ARNT, described vide infra, and developed computational/analytical protocols that can be utilized to assess additional pathological variants in future studies. Many structural studies are static, e.g., investigating an X-ray crystal structure. While such information is invaluable, it does have limitations and caveats. The crystal growth and data collection conditions are typically exceedingly artificial, generally with high salt and protein concentrations, extremely viscous, and at cryogenic temperatures. This may result in artificial interactions that can obfuscate the biologically relevant ones. Furthermore, proteins function dynamically, not statically, and a complete understanding of them cannot be achieved in a time-independent examination. With our MDS, we are able to replicate physiological conditions and model how single point mutations can have large effects on the conformations and interactions of proteins, in the context of their innately dynamic natures.
The specific variants we investigated were T46R, D707H, G715V, and D740H. Residue 46 is located in the bHLH domain and interfaces with ARNT. Variant T46R shows severe loss of activity in reporter assays with both the ubiquitous ARNT1 and neuron-enriched ARNT2, and is associated with obesity, though patients lack other PWL symptoms [
6]. The remaining three mutants all reside in the SIM C-terminal domain, which, by homology, is hypothesized to be involved with SIM1’s transcriptional regulatory activity [
7]. D707H had a moderate impact on transcriptional activity with ARNT1 and ARNT2 and was associated with obesity [
8]. Similarly, G715V had a moderate impact on activity with ARNT1 and ARNT2 and was associated with obesity in at least two unrelated patients [
9]. D740H actually had a modest increase in activity with ARNT1 but not ARNT2; patients presented with obesity, but no other PWL symptoms [
6]. We chose these variants in order to discern the tripartite relationship between transcriptional activation (functional), the associated SIM1 variants (structural), and time-dependent behavior of the proteins (dynamical).
3. Results
A complete full-length, 766 amino acid structural model for human SIM1 protein was generated. The protein consists of the bHLH, PASA, PASB, PAC, and single-minded C-terminal domains, which are covered by residues 1–53, 77–147, 218–288, 292–335, and 336–766, respectively (
Figure 1A). The gap regions formed from residues 54–76, 148–217, and 289–291 are loop-rich and have low homology to catalogs of existing structures. Our structural methods have been previously described [
27,
38,
39,
40]. We implemented our modeling technologies [
27,
38,
39,
40] to achieve the first all-atom structural model for SIM1 dimerized with the ARNT crystal structure (
Figure 1B,C). The ARNT structure was reported at 2.63 Å resolution for residues 98–142, 159–228, 259–271, 301–315, 334–346, and 360–464, with gaps in the reported PDB for residues 143–158, 229–258, 272–300, 316–333, and 347–359, respectively [
41]. Our ARNT structure is based solely on the available X-ray structures and used to generate contact data for the ARNT: SIM1 heterodimer (
Figure 1B,C). Molecular dynamics simulations were completed on several variants of SIM1 with known pathogenicity (T46R, D707H, G715V, and D740H) (
Figure 1A). Inset images on
Figure 1B,C reveal the top and side view for SIM1 in its apo form.
3.1. SIM1 and ARNT Heterodimer Interface Has Contacts Consistent with Stabilizing Interface
We observed numerous strong contacts between ARNT and the SIM1 heterodimer structure. Additionally, we found many soft contacts that contribute favorably to the interface. For strong contacts, the H-bond 2.2–2.5 Å and 20° angle is considered strong, while 2.6–3.2 Å is considered a soft contact. The default is set at 2.5 Å for our cutoff, which is technically a strong contact.
Section 3.1.1. through
Section 3.1.5. discuss a domain-by-domain analysis of the dimer contacts.
3.1.1. SIM1 bHLH Domain
For the SIM1 bHLH domain, ARNT forms contacts for strong interaction pairs between ARNT: R100 side chain nitrogen (NH1) to SIM1:Glu16 side chain oxygen (O
ε2), ARNT: E98 backbone nitrogen (N
H1) to SIM1:E19 side chain oxygen (O
ε2), and ARNT: R101 side chain nitrogen (N
H1) to SIM1:D38 side chain (O
δ2). While, the soft contacts for the SIM1 bHLH domain of the ARNT:SIM1 heterodimer include ARNT residues E98, R99, R100, R101, R102, N103, K104, M105, T106, Y108, E111, L112, D114, M115, and V116, and SIM1 residues R12, E14, E16, N17, S18, E19, F20, L23, A24, L27, D38, K39, A40, I42, I43, L45, T46, and L50, respectively (
Figure 1B,C and
Figure 2A).
3.1.2. SIM1 PASA Domain
The SIM1 PASA domain forms strong interactions with ARNT via ARNT: E163 side chain oxygen (O
ε1) to SIM1:H83 side chain nitrogen (N
δ2), and ARNT: S141 side chain oxygen (O
g) to SIM1:E79 side chain oxygen (O
ε2). Soft contacts for the SIM1 PASA domain of the ARNT:SIM1 heterodimer include ARNT residues P117, T118, A121, L122, H138, M139, K140, S141, L142, R143, L159, T160, Q162, E163, H166, L167, A171, A172, D191, S192, V193, T194, P195, V196, and N198, and SIM1 residues W64, G65, H66, L73, N75, R78, E79, L80, G81, S82, H83, L84, Q86, T87, L88, E106, T107, and Q116, respectively (
Figure 1B,C and
Figure 2B).
3.1.3. SIM1 PASB Domain
For the SIM1 PASB domain, strong contacts occur between ARNT: R278 side chain (N
H1) to SIM1:P449 backbone carbonyl oxygen (O). ARNT forms soft contacts with the SIM1 PASB domain through ARNT residues I364, S365, R366, F375, V376, D377, H378, Q387, I458, F446, Q447, N448, P449, and Y450, and SIM1 residues H267, H270, G271, C272, T274, F275, and R278, respectively (
Figure 1B,C and
Figure 2C).
3.1.4. SIM1 PAC Domain
ARNT forms strong interaction pairs with the SIM1 PAC domain between ARNT: E362 side chain (O
ε2) to SIM1:P449 backbone carbonyl oxygen (O), and ARNT: D377 side chain (O
d2) to SIM1:W304 side chain (N
ε1). While, the ARNT:SIM1 PAC domain soft contacts include ARNT residues S259, R260, R261, S262, F263, I264, H307, C308, T309, G310, and Y311, and SIM1 residues R296, G302, G303, W304, Q308, Y310, A311, T312, V314, H315, N316, S317, R318, V326, S327, Y330, and T335, respectively (
Figure 1B,C and
Figure 2C,D).
3.1.5. SM Domain of SIM1 and ARNT
The SM domain of SIM1 and ARNT possess strong interaction pairs among ARNT:E223 side chain oxygens O
ε1 and O
ε2 to SIM1:T481 side chain (O
g1) and SIM1:Q753 side chain (N
ε2) respectively, ARNT:S259 side chain oxygen (O
g) (via hydrogen) to SIM1:E336 side chain (O
ε1), and ARNT:D219 side chain (O
δ2) to SIM1:Y447 side chain hydroxyl (O
H). While, the soft contacts for SIM1 SM domain of the heterodimer include ARNT residues D219, G258, K220, R222, Q224, S226, M255, C256, M257, and E312, and SIM1 residues E336, G480, T481, Y477, A752, Q753, H755, K756, and G757, respectively (
Figure 1B,C and
Figure 2D). These contacts are given in greater mechanistic detail in the following section on analysis measurements (
Figure 3) and in the
Supplemental Materials.
3.2. SIM1 Dynamics Demonstrates Pathogenic Variants Have Both Local and Global Effects
The two most illustrative poses for SIM1 are shown in
Figure 4A (namely, side and top view). The placement of the pathogenic variants is either on the far N-terminal side (position 46) or well into the C-terminal end of the SM domain (positions 707, 715, 740) (
Figure 4A–E). A simulation for the full-length wild-type (WT) SIM1 (apo) was completed (
Supplementary Movie S1). Simulations were performed on apo SIM1 to assess the impact that the variant-mutation would induce on the conformational dynamics prior to association with ARNT. Variations in the conformational presentation would thereby reduce the likelihood of proper SIM1: ARNT interface. We believe a mechanistic investigation of the variants would be biased by the presence of ARNT pre-dimerized with SIM1 in the simulations, failing to reveal important structural reorganizations influencing the interface. The central region (residues 300–335) is not displayed here, in order to reveal interesting possible interactions between the N-terminal regions and C-terminal regions of SIM1 in its apo form (
Figure 5 and
Supplementary Movie S1). Global measurements are given and discussed in
Section 3.2.1. through
Section 3.2.4.
3.2.1. T46R Variant
T46R variant engenders some reorganization, likely due to replacing the polar uncharged threonine to the positively charged arginine, which has a bulky guanidinium moiety (
Figure 4B). Adjacent residues K51, L50, Y49, M52, Y21 K25, I43, R44, and T47 are all affected over the 150 ns simulation by the T46R mutation (
Figure 4B and
Supplementary Movies S2 and S3). The
Supplementary Movies show a side-by-side comparison of the first 300 aa from the WT and R46 SIM1 protein and a zoom into the region within 12 Å of T46. Compared to the other variants, it has the least amount of local alteration.
The next set of variants covers the single-minded C-terminal domain and was simulated for >150 ns (
Supplementary Movie S4).
Supplementary Movie S4 shows WT versus all four variants in the SM domain, T46R, D707H, G715V, and D740H.
3.2.2. D707H Variant
The D707H variant induces reorganization via substituting negatively charged Asp to positively charged His (
Figure 4C). Nearby residues R525, F699, H702, Y705, F706, H707, K708, H709, Y711, and T712 are all disturbed over the 150 ns simulation by the change from the D to H variant (
Figure 4C and
Supplementary Movie S5). The
Supplementary Movie shows a zoom into the 12 Å region surrounding H707 during the simulation.
3.2.3. G715V Variant
The G715V variant propagates reorganization due to the insertion of a hydrophobic moiety (valine) where there was no side chain (
Figure 4D). Adjacent residues R521, H523, R525, T711, L713, T714, V715, Y716, and H720 are all influenced over the 150 ns simulation by the change from the G to V variant (
Figure 3A and
Figure 4D, and
Supplementary Movie S5). The
Supplementary Movie exhibits a close-up of the 12 Å region surrounding V715 during the simulation. This valine could be particularly upsetting to the helix arrangement (
Figure 3A).
3.2.4. D740H Variant
The D740H variant effectuates reorganization likely due to exchanging negatively charged Asp to positively charged His (
Figure 4E). Proximate residues A474, N511, S512, P514, I682, N729, Y730, L732, H738, and F739 are all impacted over the 150 ns simulation by the change from the D to H variant (
Figure 4E and
Supplementary Movie S5). The
Supplementary Movie displays the 12 Å region surrounding H740 during the simulation.
3.3. Detailed Analyses for Local Deviations in Geometry Lead to Larger Amplitude Changes via Correlated Motions
The global measure of the change in the entire full-length SIM1 (apo) during molecular dynamics simulations is given (
Figure 6). Here, we report that only G715V has a more grossly changed state over the course of the 150 ns simulation. However, we observed interesting apo SIM1 WT motion between N-term and C-term (
Figure 5A,B, and
Supplementary Movie S1), which could be an unbound stable form of SIM1. Mutant G715V gives RMSD around 15 Å from the starting conformation, having gone into completely different global orientation between the N- and C-term due to the flexibility from the loosened helix. Mutants T46R and D707H showed less RMSD shift than WT (~5 Å difference), while mutant D740H converges with WT after 100 ns of simulation (
Figure 4). The global RMSD does show that after approximately 100 ns, each of the simulations settled to a general global conformation, as the RMSD only retains small fluctuations around an average. The initial structure for all of these models was essentially the same, aside from in silico point mutations, and each variant settled to a different average RMSD with respect to this initial state: G715V~15 Å, WT~12 Å, D740H~11 Å, T46R~9 Å, and D707H~9 Å. Because the global comparisons do not address individual residues or other reasons for variants’ loss-of-activity, we pursued multiple other metrics for analysis.
First, local RMSD within 8 Å of the mutant gives a good indication of how much local geometric rearrangement occurs as a consequence of the individual variant, which were measured with respect to the initial frame from the WT structure for SIM1. Mutant T46R has the smallest RMSD change from the set (~3 Å from initial), while H707, V715, and H740 all have large jumps to >6 Å from their initial frames (
Figure 3B). Mutant D740 has the biggest change early in the simulation and then the residues settle around 8 Å from initial, while V715 shows the greatest number of large fluctuations ranging from 2–10 Å for the first 45 ns, which corresponds to the helix loosening and destabilization of the SM domain. The H707 mutant has much the same effect as the H740, but lesser amplitude (~6 Å average).
To further assess the effect of the variants on the rest of the structure, a root-mean-square-fluctuation (RMSF) per residue calculation was completed to determine which residues fluctuated the most over the entire time course of the simulation, i.e., time-averaged fluctuation (
Figure 3C). While flattened values indicate a region of lower mobility, the larger fluctuating residues indicate a more dynamic structure undergoing rapid changes that can contribute to large conformational changes. Ignoring the trailing tail ends (1–20 and 750–766) is generally prudent when considering RMSF, since the termini unsurprisingly have mobility in excess of other regions of the protein.
Mutant H740 had the largest amplitude changes (6–13 Å), which were in residues 65–75, 150, 169, 199, 338–362, 423–431, 451–452, 556, 689, and 735 (
Figure 3C). D740 is located at a partially buried sheet that is tightly neighboring nearby residues. D740 appears optimized in that position, with strong polar contacts to R471, S731, and N729. A histidine in that position makes severe clashes with those and/or other residues, and is also likely repelled by R471. To harbor a histidine, the extensive reordering of nearby regions is evidenced via the various substantial RMSF peaks in proximity to the aforementioned polar contacts. Close behind in amplitude (4–11 Å) was mutant V715, which occurred in residues 43–66, 105–107, 114, 155, 198, 257, 343–368, 406–435, 480, 535–581, 637–638, and 735–742. G715 is located at the partially buried side of a helix, and a valine in that position makes severe clashes with the side chains of Y711 and E719, as well as the backbone and C-beta of H523. The significant structural rearrangements that need to occur to accommodate a valine in that semi-buried portion of helix corroborate the largest global RMSD and significant localized RMSF peaks of that variant. Mutant H707 has only a few peaks (>6 Å) that exceed the WT graph, which occurs at positions 57–76, 144–147, 208, and 545. D707 resides on a helix with nearby residues Q704, K708, R525, and H527, notably mainly basic residues. Interesting, R46 mostly mirrors WT RMSF, but does have a few peaks with different values: position 63–65 (8.84 Å) surpasses WT (~6 Å) and 532–603 is lower than WT by ~2 Å for that entire sequence, and similarly, stays flattened from 682–740. T46 is on the solvent-exposed side of a helix, with fewer residues within reach of the sidechain, and exchanging from a polar to a charged residue in a solvent-exposed and unconstrained area explains the minimal dynamical impact, shown via minimal RMSD and RMSF changes. However, T46 is one of the bHLH residues that makes direct contact with ARNT, therefore an exchange to a bulkier residue arginine is likely the etiology of the pathogenicity of this mutant. In general, regions of elevated RMSF in the variants often correspond to portions of domains that make direct contact with ARNT, as discussed in
Section 3.1. The majority of these elevated peaks are not in proximity to the mutations, and therefore are impacted differentially from the motions of the WT SIM1 through an allosteric mechanism, ergo most of the deleterious effects of these variants would not be uncovered via static structural predictions, perhaps except T46R.
3.4. Particle Size Changes as a Consequence of the Variant Chosen
Another typical analysis is to examine the global structure’s spatial arrangement or state of compactness. Using a radius of gyration (RoG) calculation (akin to a hydrodynamic radius), we estimate the average distance from the centroid (particle center of mass) to the edges for all atoms in the structure. The RoG can grow or shrink depending on a variety of factors.
Based on earlier observations of the interaction between the N- and C-term from apo SIM1 (
Supplementary Movie S1 and
Figure 5), it is not surprising to expect that the RoG would collapse over the course of the simulation when plotted versus time. This is precisely what is seen with RoG for the WT sequence (green line) (
Figure 3D), which seems to start around 41 Å and stabilize at ~38 Å. Variants R46, H707, and H740 maintain a larger RoG than WT but H740 does collapse to around 40 Å after 55 ns of simulation. However, both R46 and H707 maintain larger RoG at 42 and 44 Å, respectively. Intriguingly, V715 collapses in to around 34 Å after just 25 ns of simulation, thus forming the most compact of the structures. Implications for these compact versus extended conformations may alter the ease of binding to partner proteins such as ARNT. The R46 mutant shows least local deformation but large global reorganization, which may be a function against its activity.
3.5. Stabilization of the Local Region Shifts through Hydrogen Bonding Network Disruptions (Triggering the Correlated Motion Cascade)
Based on the understanding of the local and global changes in structure, examining the shift in the local hydrogen-bonding (H-bond) network versus the WT sequence can be informative for establishing a triggering mechanism that released the conformational change. All H-bonds were measured within an 8 Å cutoff of the residue (and included the entire residue within that cutoff).
Looking at the H-bonds from WT versus R46 reveals an important difference, namely the loss of over 50% of the stabilizing H-bonds (dropped from 14 to 6 H-bonds) (
Figure 3E). This loss of H-bonds could explain how the loosened N-term would maintain a larger RoG but still have smaller peaks on RMSF, since it is more unwound but not as interactive as in the C-term variants (
Supplementary Movies S2 and S4). Mutant H707 has slightly increased total average number of hydrogen bonds between 8 and 10, whereas WT in the same region is only 4–7. The V715 mutant has a similar trend to H707 with an average just over 10 and WT at 8. Mutant H740 has over 11 H-bonds on average and WT in the same region during the simulation maintains approximately 8. From this list, we can observe that R46 lost 50% while the other variants gained 20–30% hydrogen bonds during the same time interval.
3.6. Apo SIM1 Has Room to Move Forming Intra-Molecular Interactions
The effect of the dampened H-bonds in R46 carried over to the intra-domain interactions (N-term to C-term) (
Figure 5A,B), where WT has domain interaction and R46 stays extended (not shown). SIM1 residues with a possibility of interaction over the course of a very long simulation include (N-term) H119, P145, Y146, H147, S148, V151, and E153, with (C-term) Q686, T687, D690, H691, P692, and R728. Chemical crosslinking could be conducted on these as a means of abrogating ARNT binding for validation. WT has intrinsic motion to move in this way (
Supplementary Movie S1), which may help facilitate binding to ARNT or other important molecules (DNA, etc.).
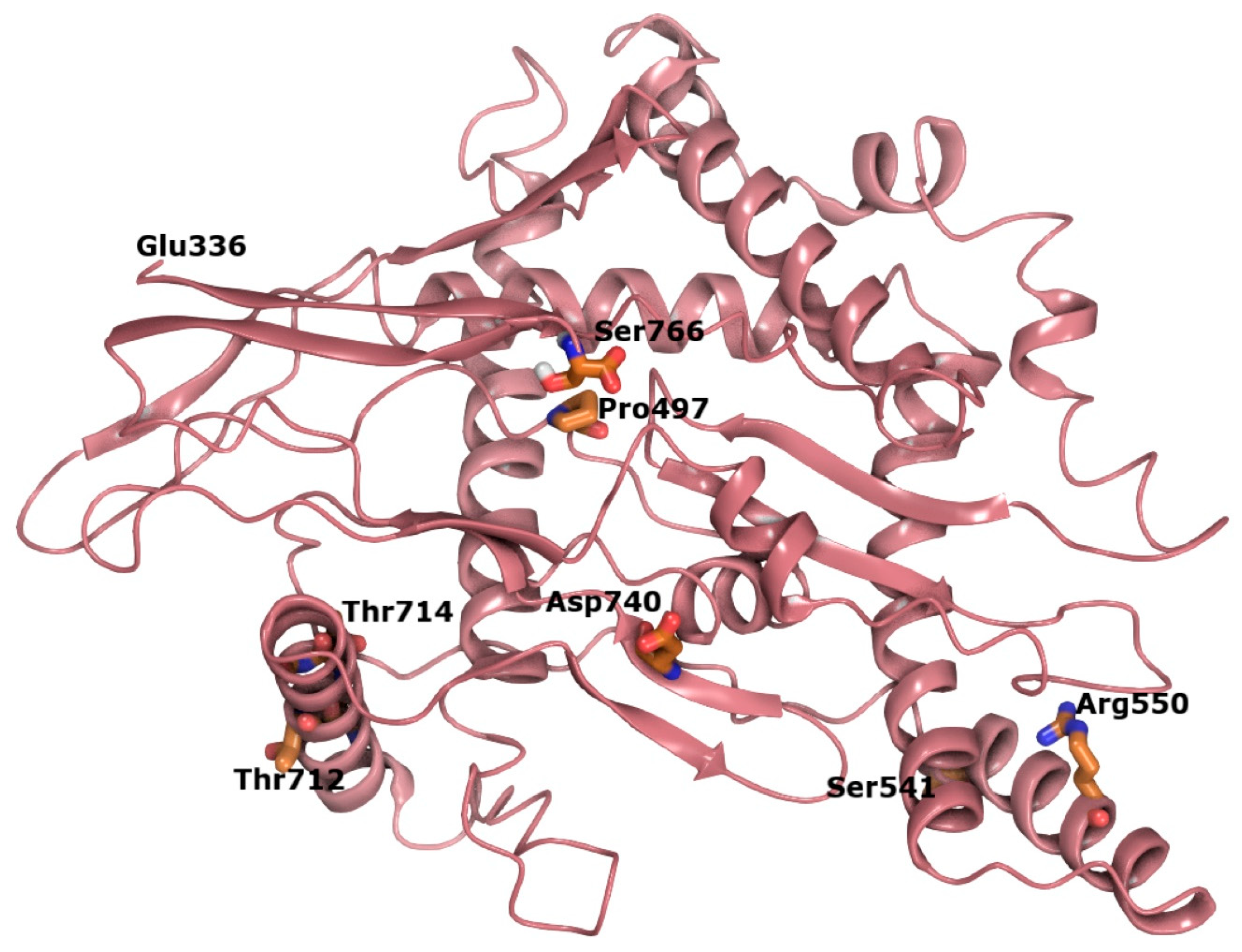
In order to examine the spatial relationship between variants in the C-terminal region of SIM1, we constructed an ab initio model of this region, which has not been structurally characterized to date (
Figure 7). The p.G715 residue is in a helix that is facing toward solvent from the protein in the single-minded 1 C-terminal domain, which has a plethora of residues as possible interactions (
Supplementary Tables S1–S3). Substitution of glycine for valine at this position leads to local increases in hydrophobicity and is predicted to disrupt helix stability over time. Validation of our hybrid model for the SIM1 through generation of a high-quality crystal structure may be useful in mapping additional variant hotspots and in generating hypotheses regarding the functional consequences of pathogenic variants that fall within the transcription regulatory domain.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}