Breakdown of Filamentous Myofibrils by the UPS–Step by Step


{kind=link}
{kind=link}
Abstract
1. Introduction
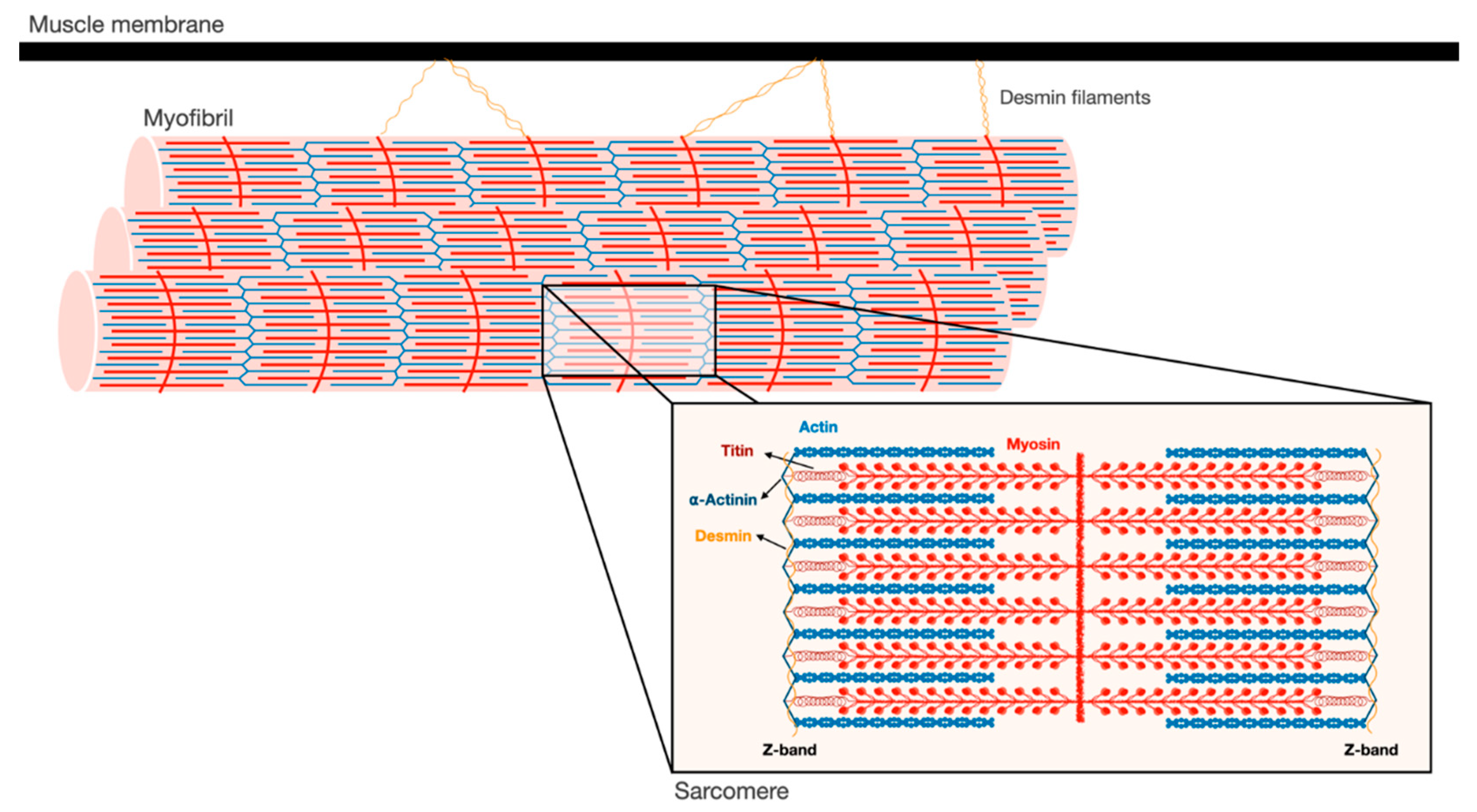
2. Myofibrils are an Intricate Filamentous Structure
3. Ubiquitin Ligases Can Act on Insoluble Filaments
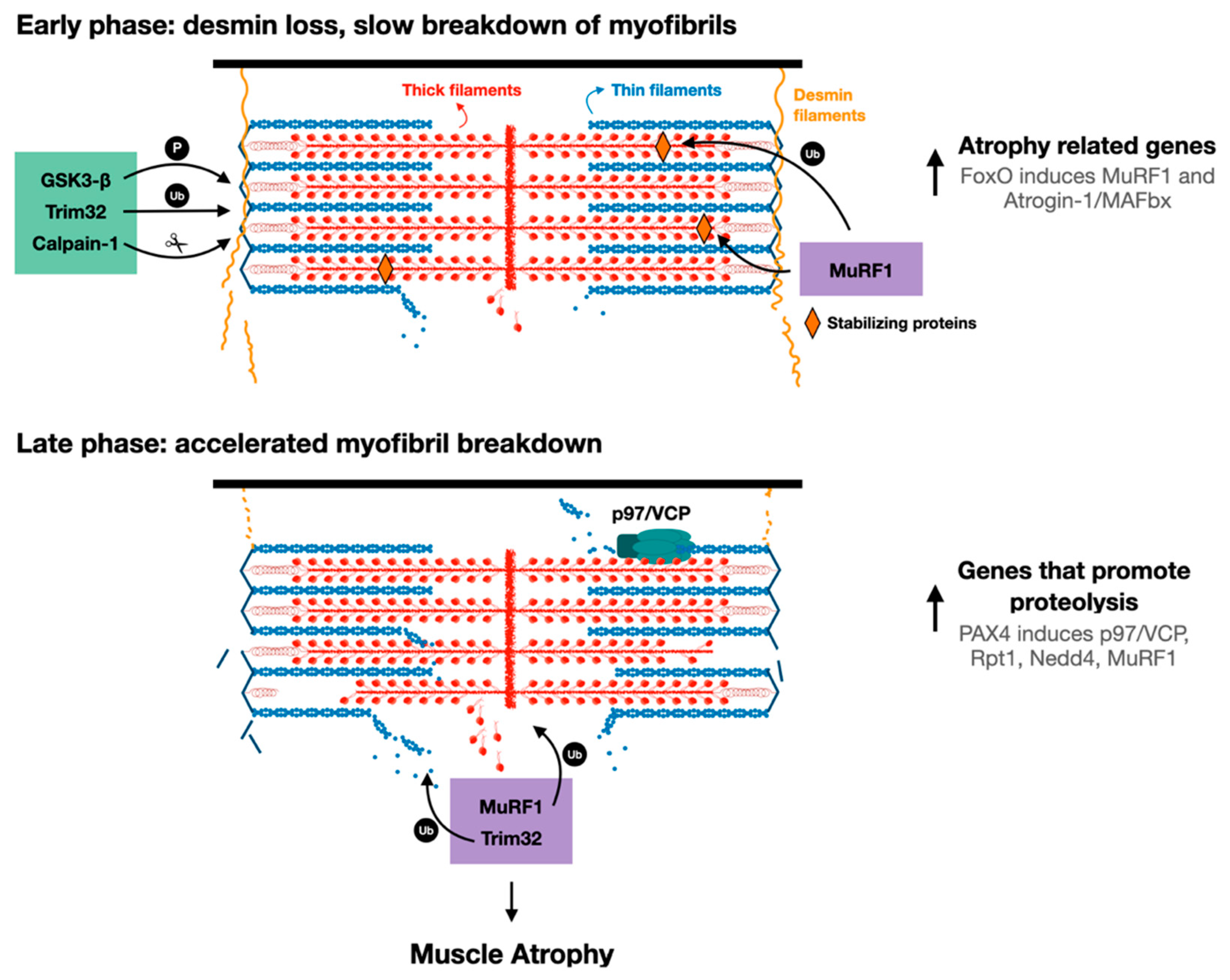
4. Loss of Stabilizing Structures is a Prerequisite to Myofibril Breakdown
5. Ubiquitinated Proteins are Released from the Myofibril by p97/VCP
6. Degradation of Myofibrillar Proteins Accompanies Systemic Disease
7. Concluding Remarks
Funding
Data Availability Statement
Conflicts of Interest
References
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nat. Cell Biol. 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.D. Protein Misfolding and Neurodegeneration. Arch. Neurol. 2008, 65, 184–189. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Lu, K.; Brave, F.D.; Jentsch, S. Pathway choice between proteasomal and autophagic degradation. Autophagy 2017, 13, 1799–1800. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.; Goldberg, A.L. Importance of the ATP-Ubiquitin-Proteasome Pathway in the Degradation of Soluble and Myofibrillar Proteins in Rabbit Muscle Extracts. J. Biol. Chem. 1996, 271, 26690–26697. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D. Skeletal Muscle Structure, Function and Plasticity. Physiotherapy 2003, 89, 565. [Google Scholar] [CrossRef]
- Aguilar, H.N.; Mitchell, B.F. Physiological pathways and molecular mechanisms regulating uterine contractility. Hum. Reprod. Updat. 2010, 16, 725–744. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, W.; Dahme, T.; Rottbauer, W.; Ackerman, M.J.; Xu, X. Depletion of zebrafish essential and regulatory myosin light chains reduces cardiac function through distinct mechanisms. Cardiovasc. Res. 2008, 79, 97–108. [Google Scholar] [CrossRef]
- Rottbauer, W.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Hassel, D.; Burns, C.G.; Katus, H.A.; Fishman, M.C. Cardiac Myosin Light Chain-2. Circ. Res. 2006, 99, 323–331. [Google Scholar] [CrossRef]
- Yang, Q.; Sanbe, A.; Osinska, H.; E Hewett, T.; Klevitsky, R.; Robbins, J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J. Clin. Investig. 1998, 102, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.A.; McElhinny, A.S.; Beckerle, M.C.; Gregorio, C.C. Striated Muscle Cytoarchitecture: An Intricate Web of Form and Function. Annu. Rev. Cell Dev. Biol. 2002, 18, 637–706. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-H.; Kuo, W.-L.; Rosner, M.R.; Tang, W.-J.; Goldman, R.D. Structural changes in intermediate filament networks alter the activity of insulin-degrading enzyme. FASEB J. 2009, 23, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Adam, S.A.; Goldman, R.D. Nuclear lamins and chromatin: When structure meets function. Adv. Enzym. Regul. 2009, 49, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef]
- Aweida, D.; Rudesky, I.; Volodin, A.; Shimko, E.; Cohen, S. GSK3-β promotes calpain-1–mediated desmin filament depolymerization and myofibril loss in atrophy. J. Cell Biol. 2018, 217, 3698–3714. [Google Scholar] [CrossRef]
- Volodin, A.; Kosti, I.; Goldberg, A.L.; Cohen, S. Myofibril breakdown during atrophy is a delayed response requiring the transcription factor PAX4 and desmin depolymerization. Proc. Natl. Acad. Sci. USA 2017, 114, E1375–E1384. [Google Scholar] [CrossRef]
- Lazarus, D.D.; Destree, A.T.; Mazzola, L.M.; McCormack, T.A.; Dick, L.R.; Xu, B.; Huang, J.Q.; Pierce, J.W.; Read, M.A.; Coggins, M.B.; et al. A new model of cancer cachexia: Contribution of the ubiquitin-proteasome pathway. Am. J. Physiol. Content 1999, 277, E332–E341. [Google Scholar] [CrossRef]
- Llovera, M.; García-Martínez, C.; Agell, N.; Marzábal, M.; López-Soriano, F.J.; Argilés, J.M. Ubiquitin gene expression is increased in skeletal muscle of tumour-bearing rats. FEBS Lett. 1994, 338, 311–318. [Google Scholar] [CrossRef]
- Pepato, M.T.; Migliorini, R.H.; Goldberg, A.L.; Kettelhut, I.C. Role of different proteolytic pathways in degradation of muscle protein from streptozotocin-diabetic rats. Am. J. Physiol. Metab. 1996, 271, E340–E347. [Google Scholar] [CrossRef] [PubMed]
- Price, S.R.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; England, B.K.; Ding, X.; Phillips, L.S.; E Mitch, W. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Investig. 1996, 98, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Tiao, G.; Fagan, J.; Roegner, V.; Lieberman, M.; Wang, J.J.; E Fischer, J.; O Hasselgren, P. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Investig. 1996, 97, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.J.; Alamdari, N.; O’Neal, P.; Gonnella, P.; Aversa, Z.; Hasselgren, P.-O. Sepsis increases the expression and activity of the transcription factor Forkhead Box O 1 (FOXO1) in skeletal muscle by a glucocorticoid-dependent mechanism. Int. J. Biochem. Cell Biol. 2010, 42, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.L.; Wang, X.; England, B.K.; Price, S.R.; Ding, X.; Mitch, W.E. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J. Clin. Investig. 1996, 97, 1447–1453. [Google Scholar] [CrossRef]
- Strassburg, S.; Springer, J.; Anker, S.D. Muscle wasting in cardiac cachexia. Int. J. Biochem. Cell Biol. 2005, 37, 1938–1947. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.-H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 Degrades Myosin Heavy Chain Protein in Dexamethasone-Treated Skeletal Muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Neti, G.; Mares, S.W.; Thompson, V.F. Myofibrillar protein turnover: The proteasome and the calpains1,2. J. Anim. Sci. 2008, 86, E19–E35. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Venkatraman, G.; Spencer, M.J. Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin–proteasome pathway. Hum. Mol. Genet. 2005, 14, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Tidball, J.G.; Spencer, M.J. Expression of a calpastatin transgene slows muscle wasting and obviates changes in myosin isoform expression during murine muscle disuse. J. Physiol. 2002, 545, 819–828. [Google Scholar] [CrossRef]
- Huang, J.; Forsberg, N.E. Role of calpain in skeletal-muscle protein degradation. Proc. Natl. Acad. Sci. USA 1998, 95, 12100–12105. [Google Scholar] [CrossRef]
- Purintrapiban, J.; Wang, M.-C.; Forsberg, N.E. Degradation of sarcomeric and cytoskeletal proteins in cultured skeletal muscle cells. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2003, 136, 393–401. [Google Scholar] [CrossRef]
- Goldbraikh, D.; Neufeld, D.; Eid-Mutlak, Y.; Lasry, I.; E Gilda, J.; Parnis, A.; Cohen, S. USP 1 deubiquitinates Akt to inhibit PI 3K-Akt-FoxO signaling in muscle during prolonged starvation. EMBO Rep. 2020, 21, e48791. [Google Scholar] [CrossRef]
- Coyne, E.S.; Bedard, N.; Wykes, L.; Stretch, C.; Jammoul, S.; Li, S.; Zhang, K.; Sladek, R.S.; Bathe, O.F.; Jagoe, R.T.; et al. Knockout of USP19 Deubiquitinating Enzyme Prevents Muscle Wasting by Modulating Insulin and Glucocorticoid Signaling. Endocrinology 2018, 159, 2966–2977. [Google Scholar] [CrossRef]
- Cohen, S.; Lee, D.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol. 2014, 204, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Ji, Y.; Zhang, X.; Wang, P.; Deng, K.; Jiang, X.; Ma, G.; Li, H.-L. Tripartite motif 32 prevents pathological cardiac hypertrophy. Clin. Sci. 2016, 130, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Signaling in Muscle Atrophy and Hypertrophy. Physiology 2008, 23, 160–170. [Google Scholar] [CrossRef]
- Agnetti, G.; Halperin, V.L.; Kirk, J.A.; Chakir, K.; Guo, Y.; Lund, L.; Nicolini, F.; Gherli, T.; Guarnieri, C.; Caldarera, C.M.; et al. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc. Res. 2014, 102, 24–34. [Google Scholar] [CrossRef]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Lin, D.I.; Barbash, O.; Kumar, K.S.; Weber, J.D.; Harper, J.W.; Klein-Szanto, A.J.P.; Rustgi, A.; Fuchs, S.Y.; Diehl, J.A. Phosphorylation-Dependent Ubiquitination of Cyclin D1 by the SCFFBX4-αB Crystallin Complex. Mol. Cell 2006, 24, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.; Concordet, J.-P.; Lassot, I.; Albert, I.; Santos, R.D.L.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 1999, 9, 207–211. [Google Scholar] [CrossRef]
- Shivanna, S.; Harrold, I.; Shashar, M.; Meyer, R.; Kiang, C.; Francis, J.; Zhao, Q.; Feng, H.; Edelman, E.R.; Rahimi, N.; et al. The c-Cbl Ubiquitin Ligase Regulates Nuclear β-Catenin and Angiogenesis by Its Tyrosine Phosphorylation Mediated through the Wnt Signaling Pathway. J. Biol. Chem. 2015, 290, 12537–12546. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yokoe, S.; Asahi, M. Phospholamban degradation is induced by phosphorylation-mediated ubiquitination and inhibited by interaction with cardiac type Sarco(endo)plasmic reticulum Ca2+-ATPase. Biochem. Biophys. Res. Commun. 2016, 472, 523–530. [Google Scholar] [CrossRef]
- Geisler, N.; Weber, K. The amino acid sequence of chicken muscle desmin provides a common structural model for intermediate filament proteins. EMBO J. 1982, 1, 1649–1656. [Google Scholar] [CrossRef]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Olivé, M.; Vicart, P.; Goebel, H.H. Intermediate Filament Diseases: Desminopathy. Cannabinoids Neuropsychiatr. Disord. 2008, 642, 131–164. [Google Scholar] [CrossRef]
- Ma, X.-W.; Li, Q.; Xu, P.-T.; Zhang, L.; Li, H.; Yu, Z.-B. Tetanic contractions impair sarcomeric Z-disk of atrophic soleus muscle via calpain pathway. Mol. Cell. Biochem. 2011, 354, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.W.; O Hasselgren, P.; Hiyama, D.T.; James, J.H.; Li, S.; Rigel, D.F.; E Fischer, J. Effect of sepsis on calcium uptake and content in skeletal muscle and regulation in vitro by calcium of total and myofibrillar protein breakdown in control and septic muscle: Results from a preliminary study. Surgery 1989, 106, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.R.; Sun, X.; Williams, A.B.; Gang, G.; Pritts, T.A.; James, H.J.; Molloy, M.; Fischer, J.E.; Paul, R.J.; Hasselgren, P.-O. Dantrolene reduces serum tnfα and corticosterone levels and muscle calcium, calpain gene expression, and protein breakdown in septic rats. Shock 2001, 15, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Bossola, M.; Muscaritoli, M.; Grieco, G.; Bonelli, G.; Bellantone, R.; Doglietto, G.; Baccino, F.; Fanelli, F.R. Anticytokine treatment prevents the increase in the activity of atp-ubiquitin- and ca2+-dependent proteolytic systems in the muscle of tumour-bearing rats. Cytokine 2002, 19, 1–5. [Google Scholar] [CrossRef]
- Turner, P.; Schultz, R.; Ganguly, B.; Steinhardt, R. Proteolysis results in altered leak channel kinetics and elevated free calcium in mdx muscle. J. Membr. Biol. 1993, 133, 243–251. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, H.M.; Qiu, Y.; Hanson, P.J.; Hemida, M.G.; Wei, W.; Hoodless, P.A.; Chu, F.; Yang, D. Coxsackievirus-Induced miR-21 Disrupts Cardiomyocyte Interactions via the Downregulation of Intercalated Disk Components. PLoS Pathog. 2014, 10, e1004070. [Google Scholar] [CrossRef] [PubMed]
- Sagar, G.D.V.; Gereben, B.; Callebaut, I.; Mornon, J.-P.; Zeöld, A.; Da Silva, W.S.; Luongo, C.; Dentice, M.; Tente, S.M.; Freitas, B.C.G.; et al. Ubiquitination-Induced Conformational Change within the Deiodinase Dimer Is a Switch Regulating Enzyme Activity. Mol. Cell. Biol. 2007, 27, 4774–4783. [Google Scholar] [CrossRef] [PubMed]
- Watkins, G.R.; Wang, N.; Mazalouskas, M.D.; Gomez, R.J.; Guthrie, C.R.; Kraemer, B.C.; Schweiger, S.; Spiller, B.W.; Wadzinski, B.E. Monoubiquitination Promotes Calpain Cleavage of the Protein Phosphatase 2A (PP2A) Regulatory Subunit α4, Altering PP2A Stability and Microtubule-associated Protein Phosphorylation. J. Biol. Chem. 2012, 287, 24207–24215. [Google Scholar] [CrossRef] [PubMed]
- Twomey, C.; Qian, S.; McCarthy, J.V. TRAF6 promotes ubiquitination and regulated intramembrane proteolysis of IL-1R1. Biochem. Biophys. Res. Commun. 2009, 381, 418–423. [Google Scholar] [CrossRef]
- Bar-Nun, S.; Glickman, M.H. Proteasomal AAA-ATPases: Structure and function. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 67–82. [Google Scholar] [CrossRef]
- Piccirillo, R.; Goldberg, A.L. The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J. 2012, 31, 3334–3350. [Google Scholar] [CrossRef]
- Kimonis, V.; Fulchiero, E.; Vesa, J.; Watts, G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: Review of a unique disorder. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 744–748. [Google Scholar] [CrossRef]
- Fang, L.; Hemion, C.; Bento, A.C.P.F.; Bippes, C.C.; Flammer, J.; Neutzner, A. Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Erzurumlu, Y.; Kose, F.A.; Gozen, O.; Gozuacik, D.; Toth, E.A.; Kirmizibayrak, P.B. A unique IBMPFD-related P97/VCP mutation with differential binding pattern and subcellular localization. Int. J. Biochem. Cell Biol. 2013, 45, 773–782. [Google Scholar] [CrossRef]
- Kustermann, M.; Manta, L.; Paone, C.; Kustermann, J.; Lausser, L.; Wiesner, C.; Eichinger, L.; Clemen, C.S.; Schröder, R.; Kestler, H.A.; et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy 2018, 14, 1911–1927. [Google Scholar] [CrossRef]
- Wang, J.; Fan, Y.; Dube, S.; Agassy, N.W.; Dube, D.K.; Sanger, J.M.; Sanger, J.W. Myofibril assembly and the roles of the ubiquitin proteasome system. Cytoskeleton 2020, 77, 456–479. [Google Scholar] [CrossRef] [PubMed]
- Janiesch, P.C.; Kim, J.; Mouysset, J.; Barikbin, R.; Lochmuller, H.; Cassata, G.; Krause, S.; Hoppe, T. The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat. Cell Biol. 2007, 9, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Blice-Baum, A.C.; Sang, T.-K.; Cammarato, A. Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function. J. Cardiovasc. Dev. Dis. 2016, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.K.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2006, 21, 140–155. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Solomon, V.; Baracos, V.; Sarraf, P.; Goldberg, A.L. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 12602–12607. [Google Scholar] [CrossRef]
- Baracos, V.; DeVivo, C.; Hoyle, D.H.; Goldberg, A.L. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am. J. Physiol. Metab. 1995, 268, E996–E1006. [Google Scholar] [CrossRef]
- Bossola, M.; Muscaritoli, M.; Costelli, P.; Grieco, G.; Bonelli, G.; Pacelli, F.; Fanelli, F.R.; Doglietto, G.B.; Baccino, F.M. Increased Muscle Proteasome Activity Correlates With Disease Severity in Gastric Cancer Patients. Ann. Surg. 2003, 237, 384–389. [Google Scholar] [CrossRef]
- Gomes-Marcondes, M.C.C.; Smith, H.J.; Cooper, J.C.; Tisdale, M.J. Development of an in-vitro model system to investigate the mechanism of muscle protein catabolism induced by proteolysis-inducing factor. Br. J. Cancer 2002, 86, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schwartz, R.J.; Waddell, I.D.; Holloway, B.R.; Reid, M.B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-κB activation in response to tumor necrosis factorα. FASEB J. 1998, 12, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A.; et al. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Mutlak, Y.E.; Aweida, D.; Volodin, A.; Ayalon, B.; Dahan, N.; Parnis, A.; Cohen, S. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hummel, R.P.; James, J.H.; Warner, B.W.; Hasselgren, P.-O.; Fischer, J.E. Evidence That Cathepsin B Contributes to Skeletal Muscle Protein Breakdown During Sepsis. Arch. Surg. 1988, 123, 221–224. [Google Scholar] [CrossRef]
- Klaude, M.; Fredriksson, K.; Tjäder, I.; Hammarqvist, F.; Ahlman, B.; Rooyackers, O.; Wernerman, J. Proteasome proteolytic activity in skeletal muscle is increased in patients with sepsis. Clin. Sci. 2007, 112, 499–506. [Google Scholar] [CrossRef]
- Hasselgren, P.-O.; James, J.; Benson, D.W.; Hall-Angerås, M.; Angerås, U.; Hiyama, D.T.; Li, S.; Fischer, J.E. Total and myofibrillar protein breakdown in different types of rat skeletal muscle: Effects of sepsis and regulation by insulin. Metabolism 1989, 38, 634–640. [Google Scholar] [CrossRef]
- Kovarik, M.; Muthny, T.; Sispera, L.; HoleČek, M. The dose-dependent effects of endotoxin on protein metabolism in two types of rat skeletal muscle. J. Physiol. Biochem. 2012, 68, 385–395. [Google Scholar] [CrossRef]
- Hobler, S.C.; Tiao, G.; Fischer, J.E.; Monaco, J.; Hasselgren, P.-O. Sepsis-induced increase in muscle proteolysis is blocked by specific proteasome inhibitors. Am. J. Physiol. Content 1998, 274, R30–R37. [Google Scholar] [CrossRef]
- Moarbes, V.; Mayaki, D.; Huck, L.; Leblanc, P.; Vassilakopoulos, T.; Petrof, B.J.; Husain, S.N.A. Differential regulation of myofibrillar proteins in skeletal muscles of septic mice. Physiol. Rep. 2019, 7, e14248. [Google Scholar] [CrossRef]
- Williams, A.B.; Decourten-Myers, G.M.; Fischer, J.E.; Luo, G.; Sun, X.; Hasselgren, P. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J. 1999, 13, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.C.S.; Figueiredo, M.J.; Campos, E.C.; Soave, D.F.; Ramos, S.G.; Tanowitz, H.B.; Celes, M.R.N. Activation of Both the Calpain and Ubiquitin-Proteasome Systems Contributes to Septic Cardiomyopathy through Dystrophin Loss/Disruption and mTOR Inhibition. PLoS ONE 2016, 11, e0166839. [Google Scholar] [CrossRef] [PubMed]
- Celes, M.R.N.; Malvestio, L.M.; Suadicani, S.O.; Prado, C.M.; Figueiredo, M.J.; Campos, E.C.; Freitas, A.C.S.; Spray, D.C.; Tanowitz, H.B.; Silva, J.S.; et al. Disruption of Calcium Homeostasis in Cardiomyocytes Underlies Cardiac Structural and Functional Changes in Severe Sepsis. PLoS ONE 2013, 8, e68809. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Linke, A.; Schoene, N.; Winkler, S.M.; Adams, V.; Conradi, S.; Busse, M.; Schuler, G.; Hambrecht, R. Functional and morphological skeletal muscle abnormalities correlate with reduced electromyographic activity in chronic heart failure. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 155–161. [Google Scholar] [CrossRef]
- Van Hees, H.W.H.; Van Der Heijden, E.H.; Ottenheijm, C.A.C.; Heunks, L.M.A.; Pigmans, C.J.C.; Verheugt, F.W.A.; Brouwer, R.M.H.J.; Dekhuijzen, P.N.R. Diaphragm single-fiber weakness and loss of myosin in congestive heart failure rats. Am. J. Physiol. Circ. Physiol. 2007, 293, H819–H828. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; VanBuren, P.; LeWinter, M.M.; Lecker, S.H.; Selby, D.E.; Palmer, B.M.; Maughan, D.W.; Ades, P.A.; Toth, M.J. Mechanisms Underlying Skeletal Muscle Weakness in Human Heart Failure. Circ. Hear. Fail. 2009, 2, 700–706. [Google Scholar] [CrossRef]
- Yancey, D.M.; Guichard, J.L.; Ahmed, M.I.; Zhou, L.; Murphy, M.P.; Johnson, M.S.; Benavides, G.A.; Collawn, J.F.; Darley-Usmar, V.M.; Dell’Italia, L.J. Cardiomyocyte mitochondrial oxidative stress and cytoskeletal breakdown in the heart with a primary volume overload. Am. J. Physiol. Circ. Physiol. 2015, 308, H651–H663. [Google Scholar] [CrossRef]
- Bouvet, M.; Dubois-Deruy, E.; Alayi, T.D.; Mulder, P.; El Amranii, M.; Beseme, O.; Amouyel, P.; Richard, V.; Tomavo, S.; Pinet, F. Increased level of phosphorylated desmin and its degradation products in heart failure. Biochem. Biophys. Rep. 2016, 6, 54–62. [Google Scholar] [CrossRef][Green Version]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Landré, V.; Rotblat, B.; Melino, S.; Bernassola, F.; Melino, G. Screening for E3-Ubiquitin ligase inhibitors: Challenges and opportunities. Oncotarget 2014, 5, 7988–8013. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tang, H.; Kou, Y.; Li, R.; Zheng, Y.; Wang, Q.; Zhou, X.; Jin, L. MG132-mediated inhibition of the ubiquitin–proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 2013, 139, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.Z.; Haroun, S.; Leblanc, E.; Trensz, F.; Guindi, C.; Amrani, A.; Grenier, G. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet. Disord. 2011, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Winder, S.J.; Lipscomb, L.; Parkin, C.A.; Juusola, M. The proteasomal inhibitor MG132 prevents muscular dystrophy in zebrafish. PLoS Curr. 2011, 3, RRN1286. [Google Scholar] [CrossRef] [PubMed]
- Araujo, K.P.C.; Bonuccelli, G.; Duarte, C.N.; Gaiad, T.P.; Moreira, D.F.; Feder, D.; Belizario, J.E.; Miglino, M.A.; Lisanti, M.P.; Ambrósio, C.E. Bortezomib (PS-341) Treatment Decreases Inflammation and Partially Rescues the Expression of the Dystrophin-Glycoprotein Complex in GRMD Dogs. PLoS ONE 2013, 8, e61367. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef]
- Neti, G.; Novak, S.M.; Thompson, V.F.; Goll, D.E. Properties of easily releasable myofilaments: Are they the first step in myofibrillar protein turnover? Am. J. Physiol. Physiol. 2009, 296, C1383–C1390. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aweida, D.; Cohen, S. Breakdown of Filamentous Myofibrils by the UPS–Step by Step. Biomolecules 2021, 11, 110. https://doi.org/10.3390/biom11010110
Aweida D, Cohen S. Breakdown of Filamentous Myofibrils by the UPS–Step by Step. Biomolecules. 2021; 11(1):110. https://doi.org/10.3390/biom11010110
Chicago/Turabian StyleAweida, Dina, and Shenhav Cohen. 2021. "Breakdown of Filamentous Myofibrils by the UPS–Step by Step" Biomolecules 11, no. 1: 110. https://doi.org/10.3390/biom11010110
APA StyleAweida, D., & Cohen, S. (2021). Breakdown of Filamentous Myofibrils by the UPS–Step by Step. Biomolecules, 11(1), 110. https://doi.org/10.3390/biom11010110