
Text Mining for Building Biomedical Networks Using Cancer as a Case Study


Abstract
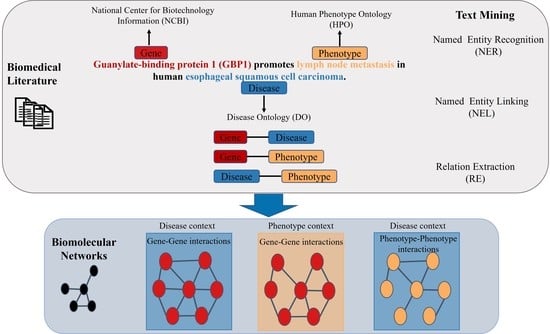
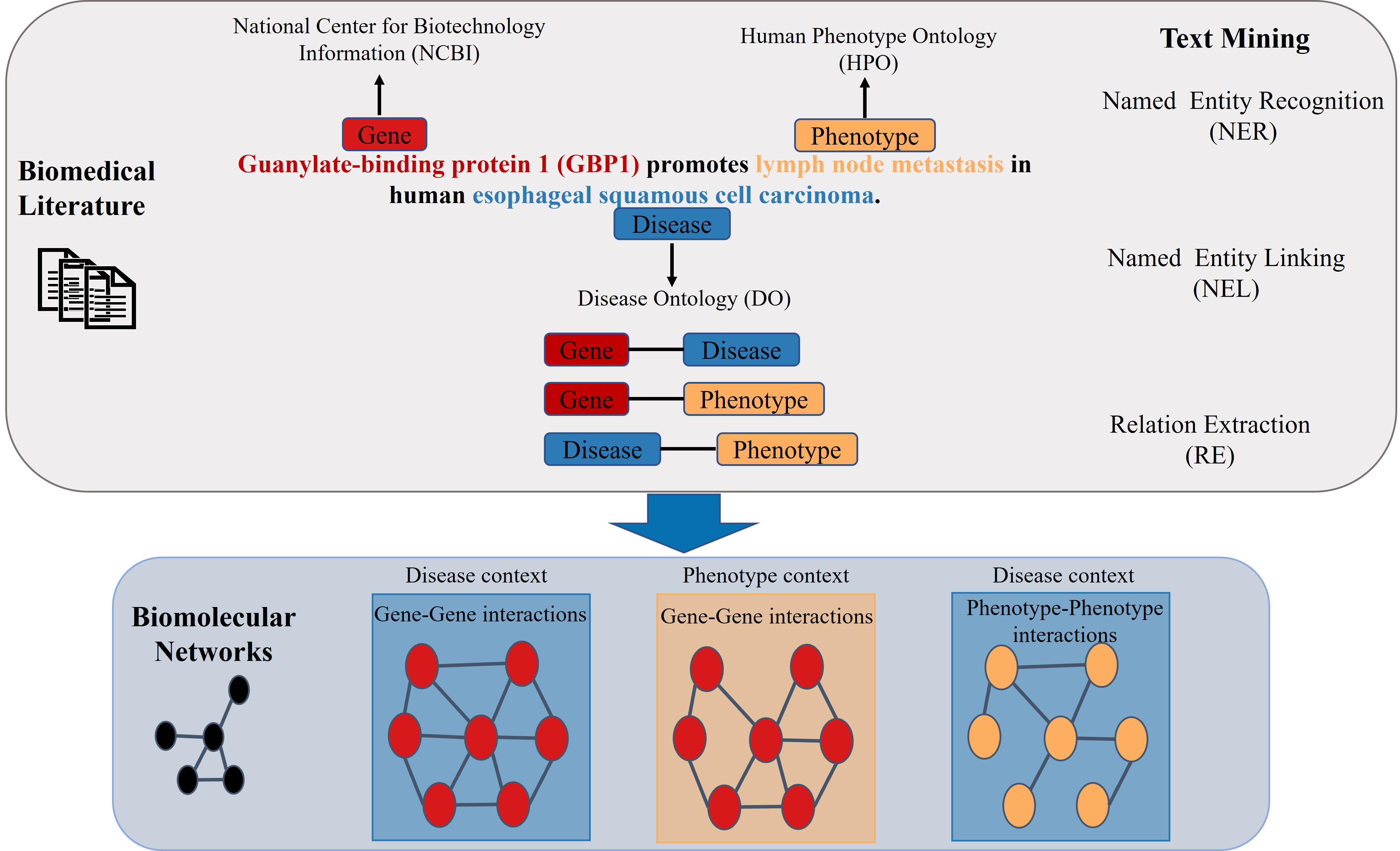
:
1. Introduction
2. Text Mining
2.1. Named Entity Recognition
2.2. Relation Extraction
- “GENE Guanylate-binding protein 1 (GBP1) promotes PHENOTYPE lymph node metastasis in human DISEASE esophageal squamous cell carcinoma.”
- 2.
- “Drug A activates the Gene A DNA repair response”
- 3.
- “Our study suggests that Drug X inhibits the function of Gene A”
2.3. Relation Extraction in the Biomedical Field
- 4.
- “The deletion mutation on exon-19 of EGFR gene was present in 16 patients, while the L858E0 point mutation on exon-21 was noted in 10. All patients were treated with gefitinib and showed a partial response.”
2.4. Databases with Text Mining Approaches
3. Cancer and Text Mining
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hwang, S.; Kim, C.Y.; Yang, S.; Kim, E.; Hart, T.; Marcotte, E.M.; Lee, I. HumanNet v2: Human gene networks for disease research. Nucleic Acids Res. 2019, 47, D573–D580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonawane, A.R.; Weiss, S.T.; Glass, K.; Sharma, A. Network medicine in the age of biomedical big data. Front. Genet. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katona, J. Analyse the Readability of LINQ Code using an Eye-Tracking-based Evaluation. Acta Polytech. Hung. 2021, 18, 193–215. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Singhal, A.; Leaman, R.; Catlett, N.; Lemberger, T.; McEntyre, J.; Polson, S.; Xenarios, I.; Arighi, C.; Lu, Z. Pressing needs of biomedical text mining in biocuration and beyond: Opportunities and challenges. Database 2016, 2016. [Google Scholar] [CrossRef]
- Lamurias, A.; Couto, F.M. Text Mining for Bioinformatics Using Biomedical Literature. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 602–611. [Google Scholar]
- World Health Organization: Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1. (accessed on 21 January 2021).
- World Health Organization: Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer. (accessed on 21 January 2021).
- Korhonen, A.; Séaghdha, D.Ó.; Silins, I.; Sun, L.; Högberg, J.; Stenius, U. Text mining for literature review and knowledge discovery in cancer risk assessment and research. PLoS ONE 2012, 7, e33427. [Google Scholar] [CrossRef]
- Zhu, F.; Patumcharoenpol, P.; Zhang, C.; Yang, Y.; Chan, J.; Meechai, A.; Vongsangnak, W.; Shen, B. Biomedical text mining and its applications in cancer research. J. Biomed. Inform. 2013, 46, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Spasic’, I.; Livsey, J.; Keane, J.A.; Nenadic’, G. Text mining of cancer-related information: Review of current status and future directions. Int. J. Med. Inform. 2014, 83, 605–623. [Google Scholar] [CrossRef]
- Couto, F.M. Data and Text Processing for Health and Life Sciences; Springer Nature: Cham, Switzerland, 2019. [Google Scholar]
- Jurca, G.; Addam, O.; Aksac, A.; Gao, S.; Özyer, T.; Demetrick, D.; Alhajj, R. Integrating text mining, data mining, and network analysis for identifying genetic breast cancer trends. Bmc Res. Notes 2016, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Habibi, M.; Weber, L.; Neves, M.; Wiegandt, D.L.; Leser, U. Deep learning with word embeddings improves biomedical named entity recognition. Bioinformatics 2017, 33, i37–i48. [Google Scholar] [CrossRef]
- Yoon, W.; So, C.H.; Lee, J.; Kang, J. Collabonet: Collaboration of deep neural networks for biomedical named entity recognition. Bmc Bioinform. 2019, 20, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Schriml, L.M.; Mitraka, E.; Munro, J.; Tauber, B.; Schor, M.; Nickle, L.; Felix, V.; Jeng, L.; Bearer, C.; Lichenstein, R.; et al. Human Disease Ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Res. 2018, 47, D955–D962. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2018, 47, D1018–D1027. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015, 44, D7–D19. [Google Scholar]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Baltoumas, F.A.; Zafeiropoulou, S.; Karatzas, E.; Paragkamian, S.; Thanati, F.; Iliopoulos, I.; Eliopoulos, A.G.; Schneider, R.; Jensen, L.J.; Pafilis, E.; et al. OnTheFly2.0: A text-mining web application for automated biomedical entity recognition, document annotation, network and functional enrichment analysis. bioRxiv 2021, 2021.05.14.444150. [Google Scholar] [CrossRef]
- Pafilis, E.; Buttigieg, P.L.; Ferrell, B.; Pereira, E.; Schnetzer, J.; Arvanitidis, C.; Jensen, L.J. EXTRACT: Interactive extraction of environment metadata and term suggestion for metagenomic sample annotation. Database 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, J.M.; Bader, G.D. Towards reliable named entity recognition in the biomedical domain. Bioinformatics 2020, 36, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, L.; Sänger, M.; Münchmeyer, J.; Habibi, M.; Leser, U.; Akbik, A. HunFlair: An easy-to-use tool for state-of-the-art biomedical named entity recognition. Bioinformatics 2021, 37, 2792–2794. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.; Couto, F.M. Knowledge representation and management: A linked data perspective. Yearb. Med. Inform. 2016, 25, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Bunescu, R.; Mooney, R.; Ramani, A.; Marcotte, E. Integrating co-occurrence statistics with information extraction for robust retrieval of protein interactions from Medline. In Proceedings of the HTLT-NAACL BioNLP Workshop on Linking Natural Language and Biology, New York, NY, USA, 8 June 2006; pp. 49–56. [Google Scholar]
- Allahyari, M.; Pouriyeh, S.; Assefi, M.; Safaei, S.; Trippe, E.D.; Gutierrez, J.B.; Kochut, K. A brief survey of text mining: Classification, clustering and extraction techniques. arXiv 2017, arXiv:1707.02919. [Google Scholar]
- Hearst, M.A. Automatic acquisition of hyponyms from large text corpora. In Proceedings of the Coling 1992 volume 2: The 14th International Conference on Computational Linguistics, Nantes, France, 23–28 August 1992; pp. 539–545. [Google Scholar]
- Deepika, S.; Geetha, T. Pattern-based bootstrapping framework for biomedical relation extraction. Eng. Appl. Artif. Intell. 2021, 99, 104130. [Google Scholar] [CrossRef]
- Mintz, M.; Bills, S.; Snow, R.; Jurafsky, D. Distant supervision for relation extraction without labeled data. In Proceedings of the Joint Conference of the 47th Annual Meeting of the ACL and the 4th International Joint Conference on Natural Language Processing of the AFNLP, Suntec, Singapore, August 2009; pp. 1003–1011. [Google Scholar]
- Yan, Y.; Okazaki, N.; Matsuo, Y.; Yang, Z.; Ishizuka, M. Unsupervised relation extraction by mining wikipedia texts using information from the web. In Proceedings of the Joint Conference of the 47th Annual Meeting of the ACL and the 4th International Joint Conference on Natural Language Processing of the AFNLP, Suntec, Singapore, 2–7 August 2009; pp. 1021–1029. [Google Scholar]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J. BioBERT: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020, 36, 1234–1240. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Gruber, T.R. A translation approach to portable ontology specifications. Knowl. Acquis. 1993, 5, 199–220. [Google Scholar] [CrossRef]
- Lamurias, A.; Sousa, D.; Clarke, L.A.; Couto, F.M. BO-LSTM: Classifying relations via long short-term memory networks along biomedical ontologies. BMC Bioinform. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, D.; Couto, F.M. BiOnt: Deep Learning using Multiple Biomedical Ontologies for Relation Extraction; European Conference on Information Retrieval; Springer: Cham, Switzerland, 2020; pp. 367–374. [Google Scholar]
- Zhang, Y.; Lin, H.; Yang, Z.; Wang, J.; Zhang, S.; Sun, Y.; Yang, L. A hybrid model based on neural networks for biomedical relation extraction. J. Biomed. Inform. 2018, 81, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Luo, Z.; Wang, S. A Hybrid Deep Learning Model for Protein–Protein Interactions Extraction from Biomedical Literature. Appl. Sci. 2020, 10, 2690. [Google Scholar] [CrossRef] [Green Version]
- Peng, N.; Poon, H.; Quirk, C.; Toutanova, K.; Yih, W.t. Cross-sentence N-ary relation extraction with graph LSTMs. Trans. Assoc. Comput. Linguist. 2017, 5, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Wang, J.; Lin, H.; Wang, X.; Yang, Z.; Zhang, Y. Biomedical cross-sentence relation extraction via multihead attention and graph convolutional networks. Appl. Soft Comput. 2021, 104, 107230. [Google Scholar] [CrossRef]
- Baltoumas, F.A.; Zafeiropoulou, S.; Karatzas, E.; Koutrouli, M.; Thanati, F.; Voutsadaki, K.; Gkonta, M.; Hotova, J.; Kasionis, I.; Hatzis, P.; et al. Biomolecule and Bioentity Interaction Databases in Systems Biology: A Comprehensive Review. Biomolecules 2021, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM® McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). Available online: https://omim.org/ (accessed on 20 July 2021).
- Pinero, J.; Queralt-Rosinach, N.; Bravo, A.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.; Rocktäschel, T.; Hakenberg, J.; Lichtblau, Y.; Leser, U. SETH detects and normalizes genetic variants in text. Bioinformatics 2016, 32, 2883–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Bravo, À.; Piñero, J.; Queralt-Rosinach, N.; Rautschka, M.; Furlong, L.I. Extraction of relations between genes and diseases from text and large-scale data analysis: Implications for translational research. BMC Bioinform. 2015, 16, 55. [Google Scholar] [CrossRef] [Green Version]
- Bundschus, M.; Dejori, M.; Stetter, M.; Tresp, V.; Kriegel, H.P. Extraction of semantic biomedical relations from text using conditional random fields. BMC Bioinform. 2008, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef]
- LIterature coNcept Knowledgebase. Available online: Hhttps://link.opentargets.io/ (accessed on 27 January 2021).
- Chatr-Aryamontri, A.; Winter, A.; Perfetto, L.; Briganti, L.; Licata, L.; Iannuccelli, M.; Castagnoli, L.; Cesareni, G.; Tyers, M. Benchmarking of the 2010 BioCreative Challenge III text-mining competition by the BioGRID and MINT interaction databases. BMC Bioinform. 2011, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef]
- Buckley, J.M.; Coopey, S.B.; Sharko, J.; Polubriaginof, F.; Drohan, B.; Belli, A.K.; Kim, E.M.; Garber, J.E.; Smith, B.L.; Gadd, M.A.; et al. The feasibility of using natural language processing to extract clinical information from breast pathology reports. J. Pathol. Inform. 2012, 3, 23. [Google Scholar]
- Yala, A.; Barzilay, R.; Salama, L.; Griffin, M.; Sollender, G.; Bardia, A.; Lehman, C.; Buckley, J.M.; Coopey, S.B.; Polubriaginof, F.; et al. Using machine learning to parse breast pathology reports. Breast Cancer Res. Treat. 2017, 161, 203–211. [Google Scholar] [CrossRef]
- Kawashima, K.; Bai, W.; Quan, C. Text Mining and Pattern Clustering for Relation Extraction of Breast Cancer and Related Genes. In Proceedings of the 2017 18th IEEE/ACIS International Conference on Software Engineering, Artificial Intelligence, Networking and Parallel/Distributed Computing (SNPD), Kanazawa, Japan, 26–28 June 2017; pp. 59–63. [Google Scholar]
- Lin, H.J.; Sheu, P.C.Y.; Tsai, J.J.; Wang, C.C.; Chou, C.Y. Text mining in a literature review of urothelial cancer using topic model. BMC Cancer 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Fabacher, T.; Godet, J.; Klein, D.; Velten, M.; Jegu, J. Machine learning application for incident prostate adenocarcinomas automatic registration in a French regional cancer registry. Int. J. Med. Inform. 2020, 139, 104139. [Google Scholar] [CrossRef]
- Weinberg, R.; Hanahan, D. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Jiang, L.; Sun, X.; Mercaldo, F.; Santone, A. DECAB-LSTM: Deep Contextualized Attentional Bidirectional LSTM for cancer hallmark classification. Knowl.-Based Syst. 2020, 210, 106486. [Google Scholar] [CrossRef]
- Baker, S.; Korhonen, A.L.; Pyysalo, S. Cancer hallmark text classification using convolutional neural networks. In Proceedings of the Fifth Workshop on Building and Evaluating Resources for Biomedical Text Mining (BioTxtM2016), Osaka, Japan, 11–16 December 2016; pp. 1–9. [Google Scholar]
- Lever, J.; Jones, M.R.; Danos, A.M.; Krysiak, K.; Bonakdar, M.; Grewal, J.K.; Culibrk, L.; Griffith, O.L.; Griffith, M.; Jones, S.J. Text-mining clinically relevant cancer biomarkers for curation into the CIViC database. Genome Med. 2019, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawad, M.; Gao, S.; Qiu, J.X.; Yoon, H.J.; Blair Christian, J.; Penberthy, L.; Mumphrey, B.; Wu, X.C.; Coyle, L.; Tourassi, G. Automatic extraction of cancer registry reportable information from free-text pathology reports using multitask convolutional neural networks. J. Am. Med. Inform. Assoc. 2020, 27, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, J.J.; Zhao, X.; Mays, J.C.; Davoli, T. Not all cancers are created equal: Tissue specificity in cancer genes and pathways. Curr. Opin. Cell Biol. 2020, 63, 135–143. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Text Mining Task | ||||
|---|---|---|---|---|
| Method | Target | NER | RE | Reference |
| LSTM 1-CRFF | Genes/proteins, chemicals, diseases, cell lines and species entity types | X | [14] | |
| CollaboNet | Gene/protein, disease and chemicals | X | [15] | |
| BioBERT | Gene-Disease and Protein-Chemical | X | [31] | |
| BO-LSTM | Drug-Drug | X | [34] | |
| BiOnt | Gene, phenotypes, disease and drugs combinations | X | [35] | |
| RNN 3 + CNN 2 | Protein-Protein and Drug-Drug | X | [36] | |
| LSTM 1 + CNN 2 | Protein-Protein | X | [37] | |
| graph LSTM 1 | Drug-gene-mutation | X | [38] | |
| graph LSTM 1 | Drug-gene-mutation | X | [39] | |
| SETH | Gene variant normalization in to dbSNP or UniProt | X | [45] | |
| Befree | Gene-disease and variant-disease | X | X | [47] |
| LHGDN | Gene-Disease | X | X | [48] |
| Link | Genes, diseases, drugs and key concepts | X | [50] | |
| Target Cancer | Method | Source | Reference |
|---|---|---|---|
| Breast | Data Mining and network analysis | Biomedical Abstracts | [13] |
| Breast | Rule Based | Pathology Reports | [55] |
| Breast | Machine Learning | Pathology Reports | [56] |
| Breast | Unsupervised Learning, Text mining and Pattern mining | PubMed Articles | [57] |
| Urothelial cancer | Latent Dirichlet Allocation and Lda2vec | PubMed Abstracts and Titles | [58] |
| Prostate adenocarcinoma | Machine Learning | Pathology Reports | [59] |
| Generic | LSTM | PubMed Abstracts | [61] |
| Generic | CNN 1 | Biomedical Abstracts | [62] |
| Generic | Supervised Learning | Full PubMed | [63] |
| Generic | Multitask CNN 1 | Pathology Reports | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conceição, S.I.R.; Couto, F.M. Text Mining for Building Biomedical Networks Using Cancer as a Case Study. Biomolecules 2021, 11, 1430. https://doi.org/10.3390/biom11101430
Conceição SIR, Couto FM. Text Mining for Building Biomedical Networks Using Cancer as a Case Study. Biomolecules. 2021; 11(10):1430. https://doi.org/10.3390/biom11101430
Chicago/Turabian StyleConceição, Sofia I. R., and Francisco M. Couto. 2021. "Text Mining for Building Biomedical Networks Using Cancer as a Case Study" Biomolecules 11, no. 10: 1430. https://doi.org/10.3390/biom11101430
APA StyleConceição, S. I. R., & Couto, F. M. (2021). Text Mining for Building Biomedical Networks Using Cancer as a Case Study. Biomolecules, 11(10), 1430. https://doi.org/10.3390/biom11101430