
Description of Joint Alterations Observed in a Family Carrying p.Asn453Ser COMP Variant: Clinical Phenotypes, In Silico Prediction of Functional Impact on COMP Protein and Stability, and Review of the Literature

 , , and
, , and {kind=link}
{kind=link}
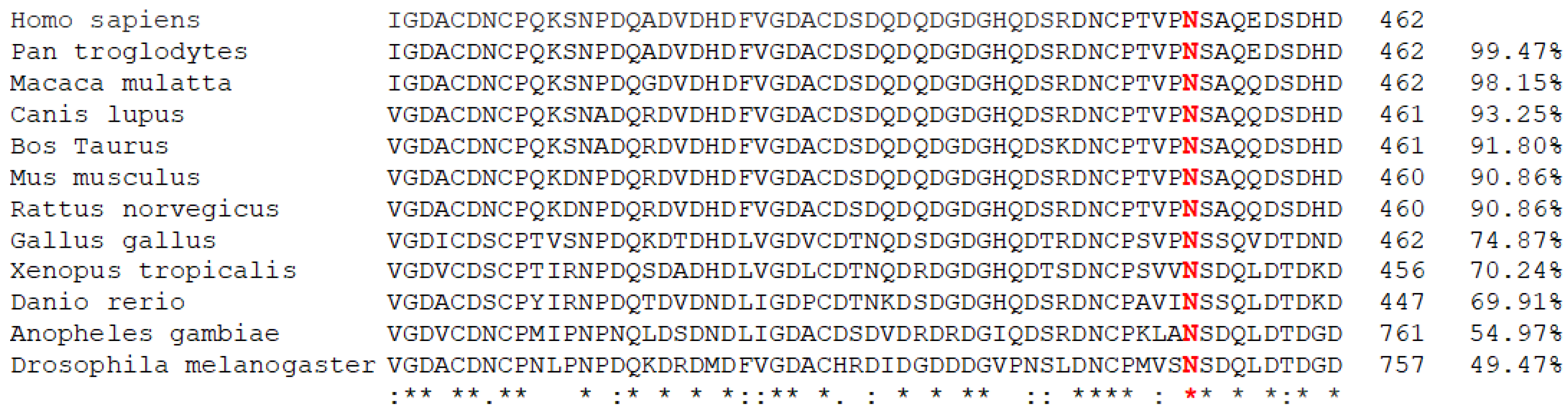{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Subjects and Methods
2.1. Subjects
2.2. Genetic Analyses
2.3. In Silico Variant Pathogenicity Prediction Analysis
2.4. Molecular Dynamics Simulations
3. Results
3.1. Clinical Presentation
3.2. Genetic Analysis and Co-Segregation
3.3. In Silico Prediction of Variation Pathogenicity
3.4. Structural Analysis and Molecular Dynamics Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandl, L.A. Osteoarthritis year in review 2018: Clinical. Osteoarthr. Cartil. 2019, 27, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Sandell, L.J. Etiology of osteoarthritis: Genetics and synovial joint development. Nat. Rev. Rheumatol. 2012, 8, 77–89. [Google Scholar] [CrossRef]
- Stecher, R. Heberden’s nodes: Heredity in hypertrophic arthritis of the finger joints. Am. J. Med. Sci. 1941, 201, 801–809. [Google Scholar] [CrossRef]
- Fernández-Moreno, M.; Rego, I.; Carreira-Garcia, V.; Blanco, F.J. Genetics in osteoarthritis. Curr. Genomics 2008, 9, 542–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aury-Landas, J.; Marcelli, C.; Leclercq, S.; Boumédiene, K.; Baugé, C. Genetic determinism of primary early-onset osteoarthritis. Trends Mol. Med. 2016, 22, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, T.D.; MacGregor, A. Risk factors for osteoarthritis: Genetics. Osteoarthr. Cartil. 2004, 12, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Sliz, E.; Taipale, M.; Welling, M.; Skarp, S.; Alaraudanjoki, V.; Ignatius, J.; Ruddock, L.; Nissi, R.; Männikkö, M. TUFT1, a novel candidate gene for metatarsophalangeal osteoarthritis, plays a role in chondrogenesis on a calcium-related pathway. PLoS ONE 2017, 12, e0175474. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wu, W.; Qu, X. Mesenchymal stem cells in osteoarthritis therapy: A review. Am. J. Transl. Res. 2021, 13, 448–461. [Google Scholar] [PubMed]
- Ballini, A.; Boccaccio, A.; Saini, R.; Van Pham, P.; Tatullo, M. Dental-Derived Stem Cells and Their Secretome and Interactions with Bioscaffolds/Biomaterials in Regenerative Medicine: From the In Vitro Research to Translational Applications. Stem Cells Int. 2017, 2017, 6975251. [Google Scholar] [CrossRef] [PubMed]
- Tatullo, M.; Spagnuolo, G.; Codispoti, B.; Zamparini, F.; Zhang, A.; Degli Esposti, M.; Aparicio, C.; Rengo, C.; Nuzzolese, M.; Manzoli, L.; et al. PLA-Based Mineral-Doped Scaffolds Seeded with Human Periapical Cyst-Derived MSCs: A Promising Tool for Regenerative Healing in Dentistry. Materials 2019, 12, 597. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; I Hurwitz, D.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2019, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; Duquette, M.; Joachimiak, A.; Lawler, J. The crystal structure of the signature domain of cartilage oligomeric matrix protein: Implications for collagen, glycosaminoglycan and integrin binding. FASEB J. 2009, 23, 2490–2501. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, G.N.M.; De Groot, B.L.; Grubmüller, S.R.B.L.D.G.H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Briggs, M.D.; Mortier, G.; Cole, W.G.; King, L.M.; Golik, S.S.; Bonaventure, J.; Nuytinck, L.; De Paepe, A.; Leroy, J.G.; Biesecker, L.; et al. Diverse Mutations in the Gene for Cartilage Oligomeric Matrix Protein in the Pseudoachondroplasia–Multiple Epiphyseal Dysplasia Disease Spectrum. Am. J. Hum. Genet. 1998, 62, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Posey, K.L.; Coustry, F.; Hecht, J.T. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 2018, 71-72, 161–173. [Google Scholar] [CrossRef]
- Otteby, K.E.; Holmquist, E.; Saxne, T.; Heinegård, D.; Hesselstrand, R.; Blom, A.M. Cartilage oligomeric matrix protein-induced complement activation in systemic sclerosis. Arthritis Res. Ther. 2013, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Acharya, C.; Yik, J.H.; Kishore, A.; Van Dinh, V.; Di Cesare, P.E.; Haudenschild, D. Cartilage oligomeric matrix protein and its binding partners in the cartilage extracellular matrix: Interaction, regulation and role in chondrogenesis. Matrix Biol. 2014, 37, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Das, B.R.; Khan, F.R.; Roy, A. Cartilage oligomeric matrix protein in monitoring and prognostication of osteoarthritis and its utility in drug development. Perspect. Clin. Res. 2015, 6, 4–9. [Google Scholar] [CrossRef]
- Mishra, A.; Awasthi, S.; Raj, S.; Mishra, P.; Srivastava, R.N. Identifying the role of ASPN and COMP genes in knee osteoarthritis development. J. Orthop. Surg. Res. 2019, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.; Reddi, A.H.; Di Cesare, P.E. Cartilage Oligomeric Matrix Protein (COMP): A Biomarker of Arthritis. Biomark. Insights 2009, 4, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Briggs, M.D.; Brock, J.; Ramsden, S.C.; Bell, P.A. Genotype to phenotype correlations in cartilage oligomeric matrix protein associated chondrodysplasias. Eur. J. Hum. Genet. 2014, 22, 1278–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styrkarsdottir, U.; Helgason, H.; Sigurdsson, A.; Norddahl, G.L.; Agustsdottir, A.B.; Reynard, L.N.; Villalvilla, A.; Halldorsson, G.H.; Jonasdottir, A.; Magnusdottir, A.; et al. Whole-genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis. Nat Genet. 2017, 49, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.-C.; Lin, Y.-J.; Liu, H.-C.; Wu, J.-Y.; Li, S.-C.; Lee, M.-T.M.; Chou, C.-H.; Chen, L.-K.; Chen, Y.-T. A Mutation in Cartilage Oligomeric Matrix Protein (COMP) Causes Early-Onset Osteoarthritis in a Large Kindred Study. Ann. Hum. Genet. 2011, 75, 575–583. [Google Scholar] [CrossRef]
- Thur, J.; Rosenberg, K.; Nitsche, D.P.; Pihlajamaa, T.; Ala-Kokko, L.; Heinegård, D.; Paulsson, M.; Maurer, P. Mutations in Cartilage Oligomeric Matrix Protein Causing Pseudoachondroplasia and Multiple Epiphyseal Dysplasia Affect Binding of Calcium and Collagen I, II, and IX. J. Biol. Chem. 2001, 276, 6083–6092. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Deere, M.; Hecht, J.T.; Lawler, J. Cartilage Oligomeric Matrix Protein Is a Calcium-binding Protein, and a Mutation in Its Type 3 Repeats Causes Conformational Changes. J. Biol. Chem. 2000, 275, 26538–26544. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Acharya, C.; Christiansen, B.A.; Yik, J.H.; DiCesare, P.E.; Haudenschild, D. Cartilage oligomeric matrix protein enhances osteogenesis by directly binding and activating bone morphogenetic protein-2. Bone 2013, 55, 23–35. [Google Scholar] [CrossRef]
- Gong, S.-D.; Chen, X.; Chen, Z.-Q.; He, X.-M.; Pang, F.-X.; Huang, J.-Y.; Zhou, Y.-C.; Qin, Y.-X.; Liu, S.-J.; Wei, Q.-S. Elevated plasma cartilage oligomeric matrix protein (COMP) level are associated with the progression of non-traumatic osteonecrosis of femoral head. Clin. Chim. Acta 2018, 490, 214–221. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochoux, Q.; Sopkova-de Oliveira Santos, J.; Marcelli, C.; Rovelet-Lecrux, A.; Chevallier, V.; Dutheil, J.-J.; Leclercq, S.; Boumédiene, K.; Baugé, C.; Aury-Landas, J. Description of Joint Alterations Observed in a Family Carrying p.Asn453Ser COMP Variant: Clinical Phenotypes, In Silico Prediction of Functional Impact on COMP Protein and Stability, and Review of the Literature. Biomolecules 2021, 11, 1460. https://doi.org/10.3390/biom11101460
Rochoux Q, Sopkova-de Oliveira Santos J, Marcelli C, Rovelet-Lecrux A, Chevallier V, Dutheil J-J, Leclercq S, Boumédiene K, Baugé C, Aury-Landas J. Description of Joint Alterations Observed in a Family Carrying p.Asn453Ser COMP Variant: Clinical Phenotypes, In Silico Prediction of Functional Impact on COMP Protein and Stability, and Review of the Literature. Biomolecules. 2021; 11(10):1460. https://doi.org/10.3390/biom11101460
Chicago/Turabian StyleRochoux, Quitterie, Jana Sopkova-de Oliveira Santos, Christian Marcelli, Anne Rovelet-Lecrux, Virginie Chevallier, Jean-Jacques Dutheil, Sylvain Leclercq, Karim Boumédiene, Catherine Baugé, and Juliette Aury-Landas. 2021. "Description of Joint Alterations Observed in a Family Carrying p.Asn453Ser COMP Variant: Clinical Phenotypes, In Silico Prediction of Functional Impact on COMP Protein and Stability, and Review of the Literature" Biomolecules 11, no. 10: 1460. https://doi.org/10.3390/biom11101460
APA StyleRochoux, Q., Sopkova-de Oliveira Santos, J., Marcelli, C., Rovelet-Lecrux, A., Chevallier, V., Dutheil, J.-J., Leclercq, S., Boumédiene, K., Baugé, C., & Aury-Landas, J. (2021). Description of Joint Alterations Observed in a Family Carrying p.Asn453Ser COMP Variant: Clinical Phenotypes, In Silico Prediction of Functional Impact on COMP Protein and Stability, and Review of the Literature. Biomolecules, 11(10), 1460. https://doi.org/10.3390/biom11101460