
Biological Profile of Two Gentiana lutea L. Metabolites Using Computational Approaches and In Vitro Tests

 ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
, 
Abstract
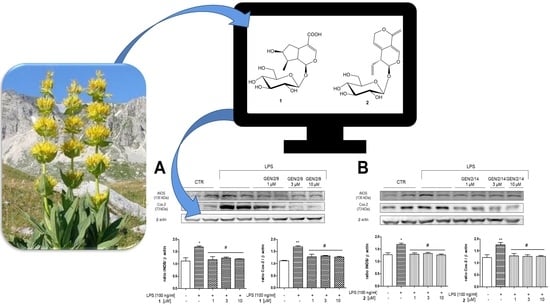
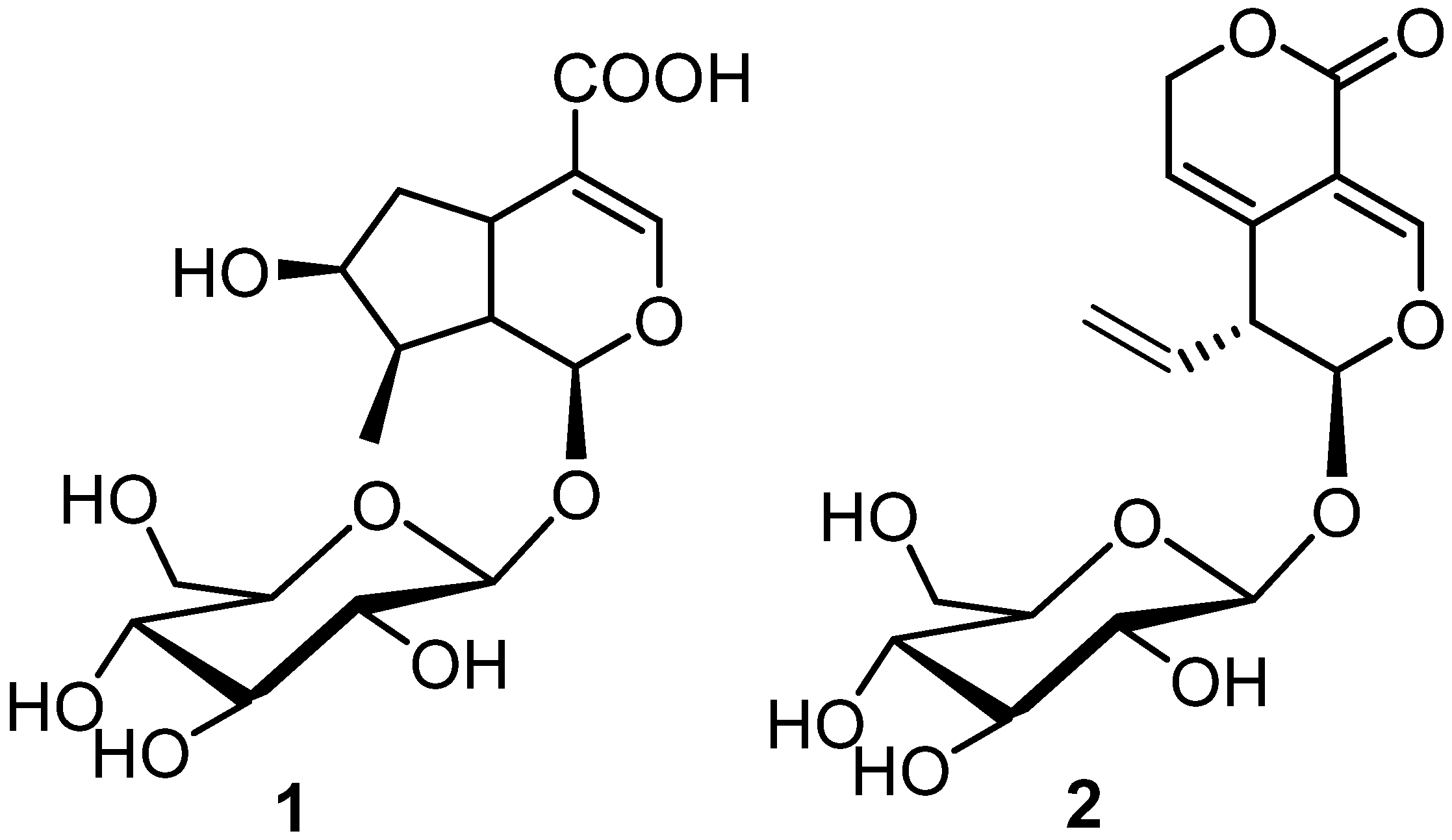
:
1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Plant Material
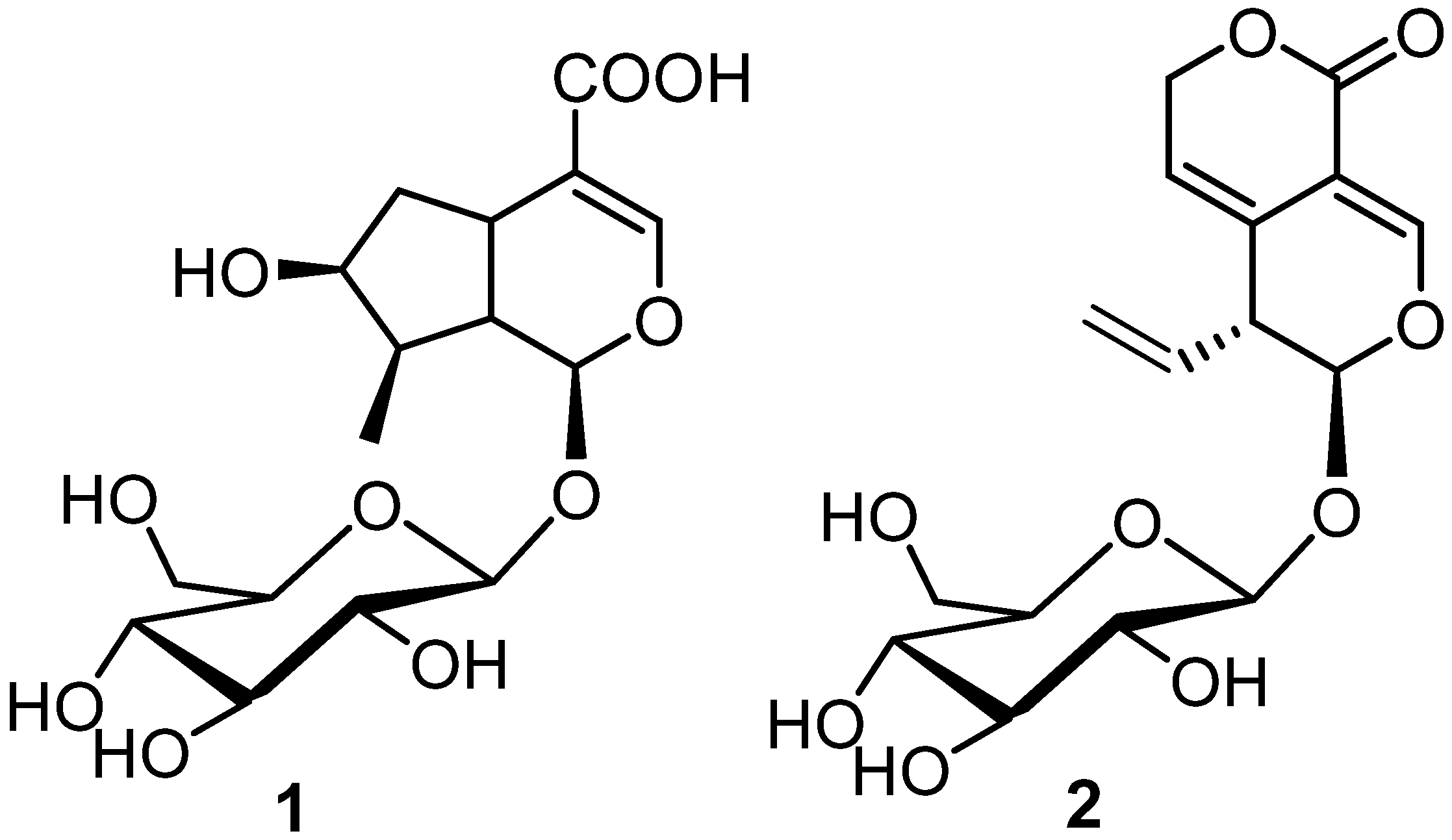
2.3. Extraction and Isolation
2.4. Inverse Virtual Screening
2.5. Input Molecules Preparation
2.6. Protein Panel Preparation
2.7. Binding Site Detection and Molecular Docking
2.8. Molecular Dynamics
2.9. In Vitro Experiments
2.9.1. Chemicals
2.9.2. Cell Culture and Treatment
2.9.3. Western Blot Analysis
2.10. Statistical Analysis
3. Results
3.1. Inverse Virtual Screening
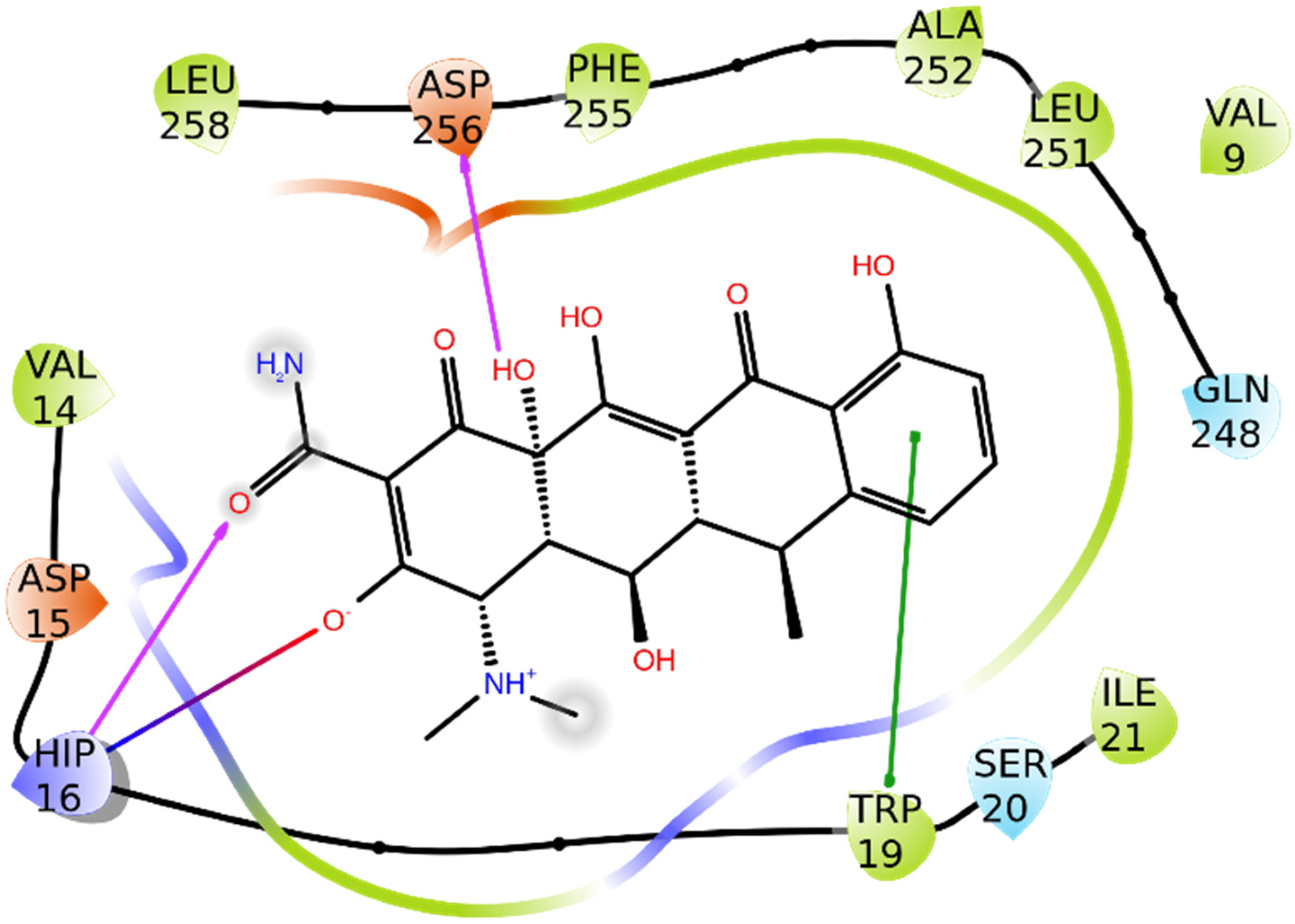
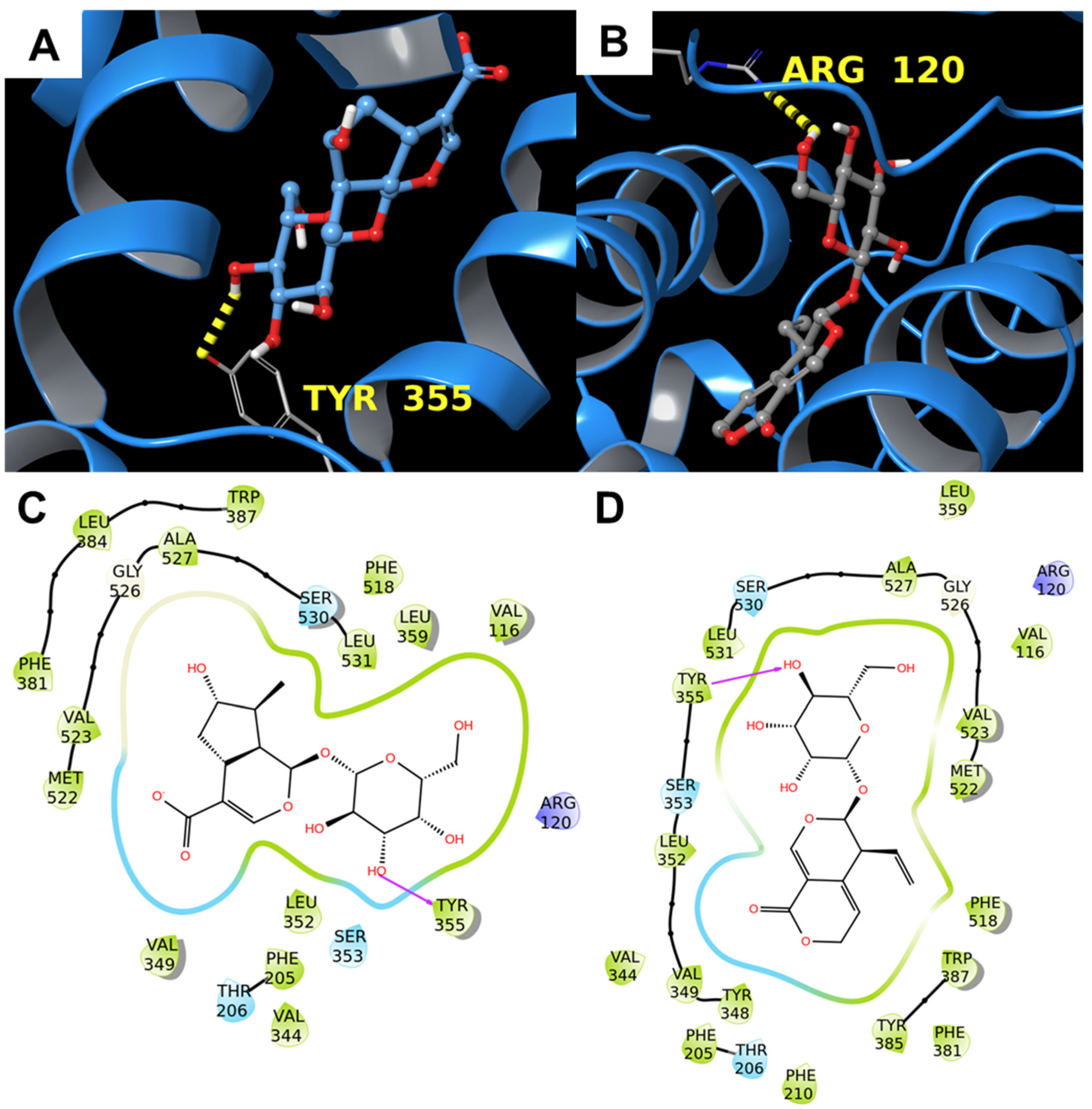
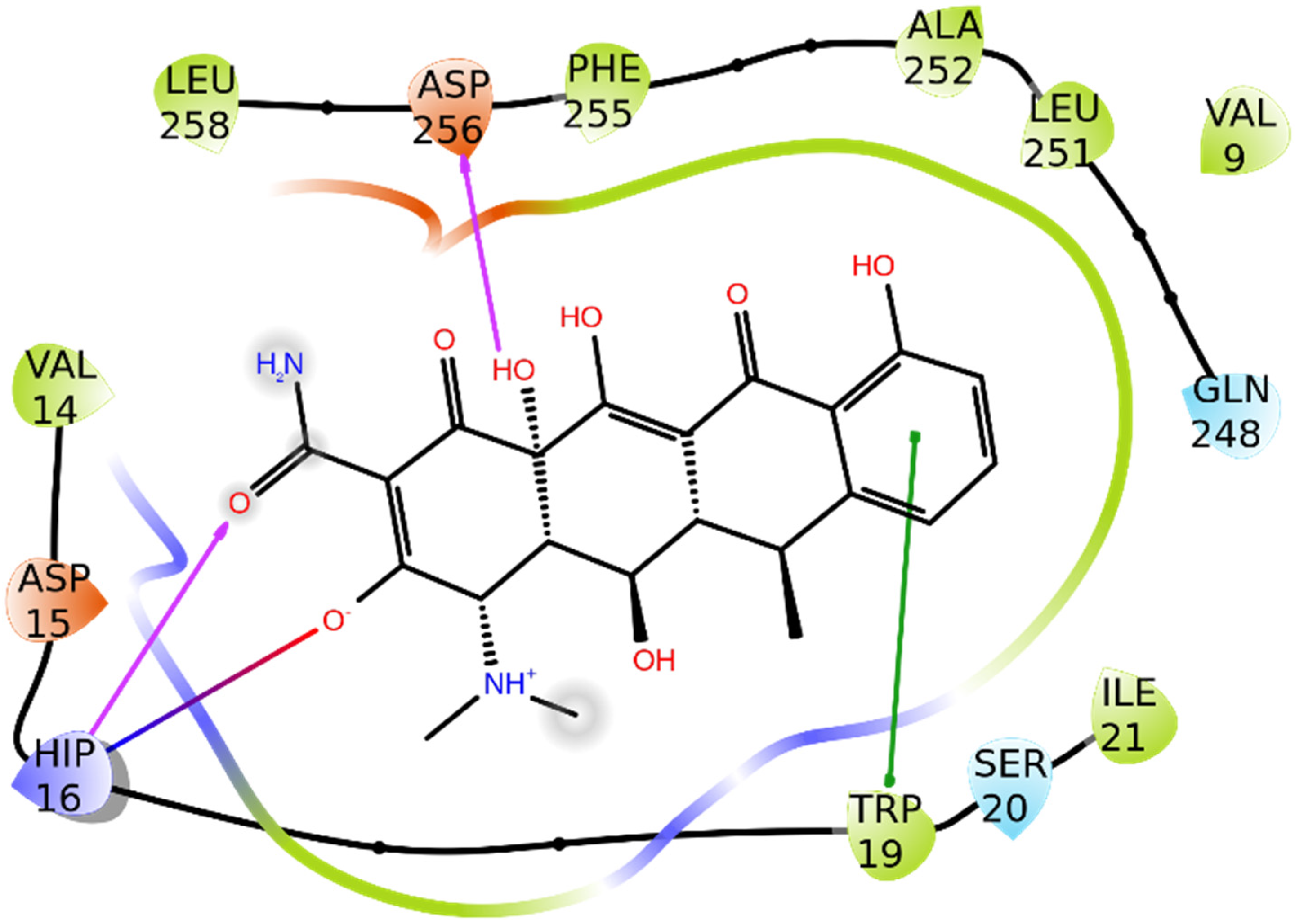
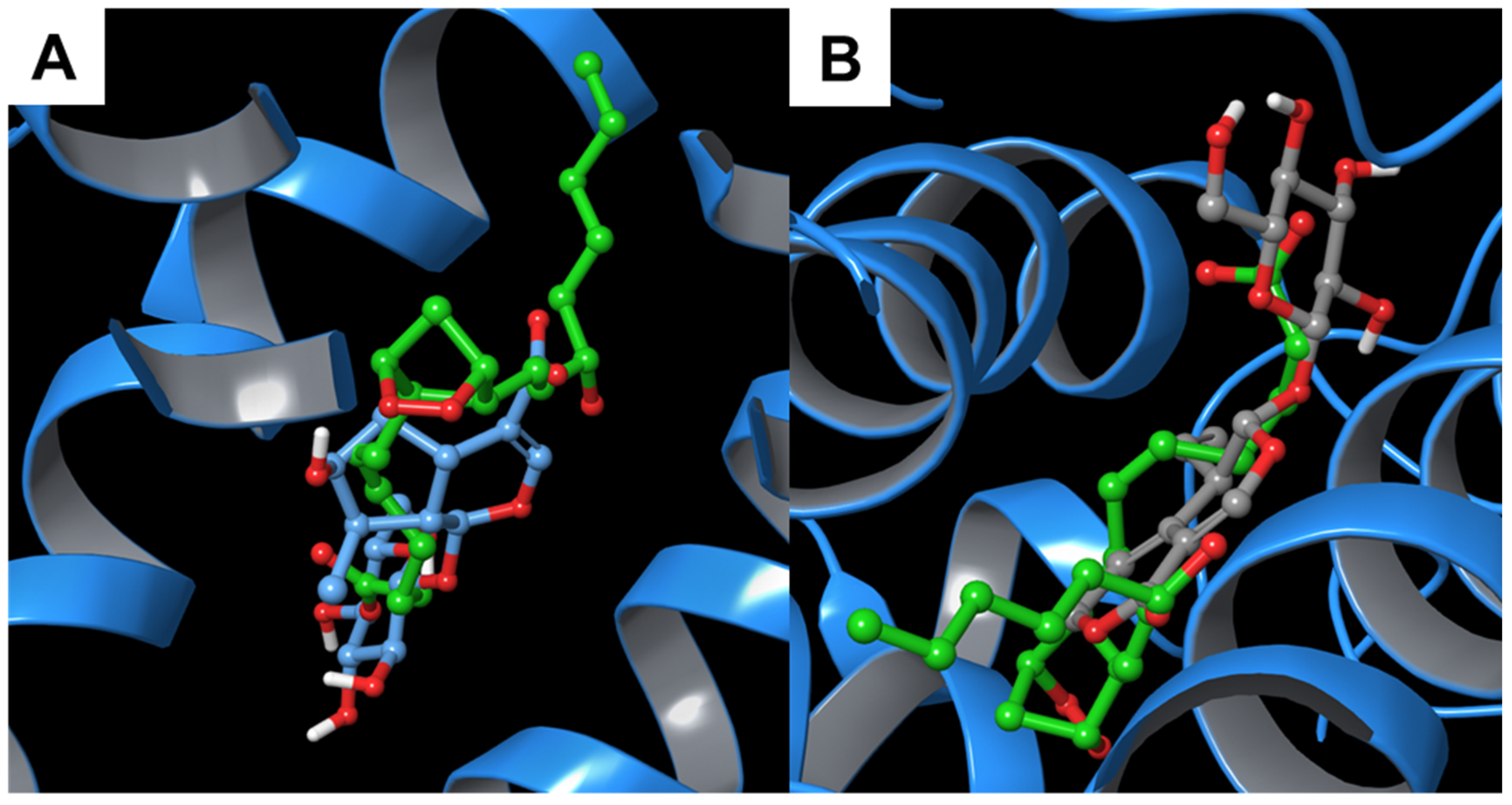
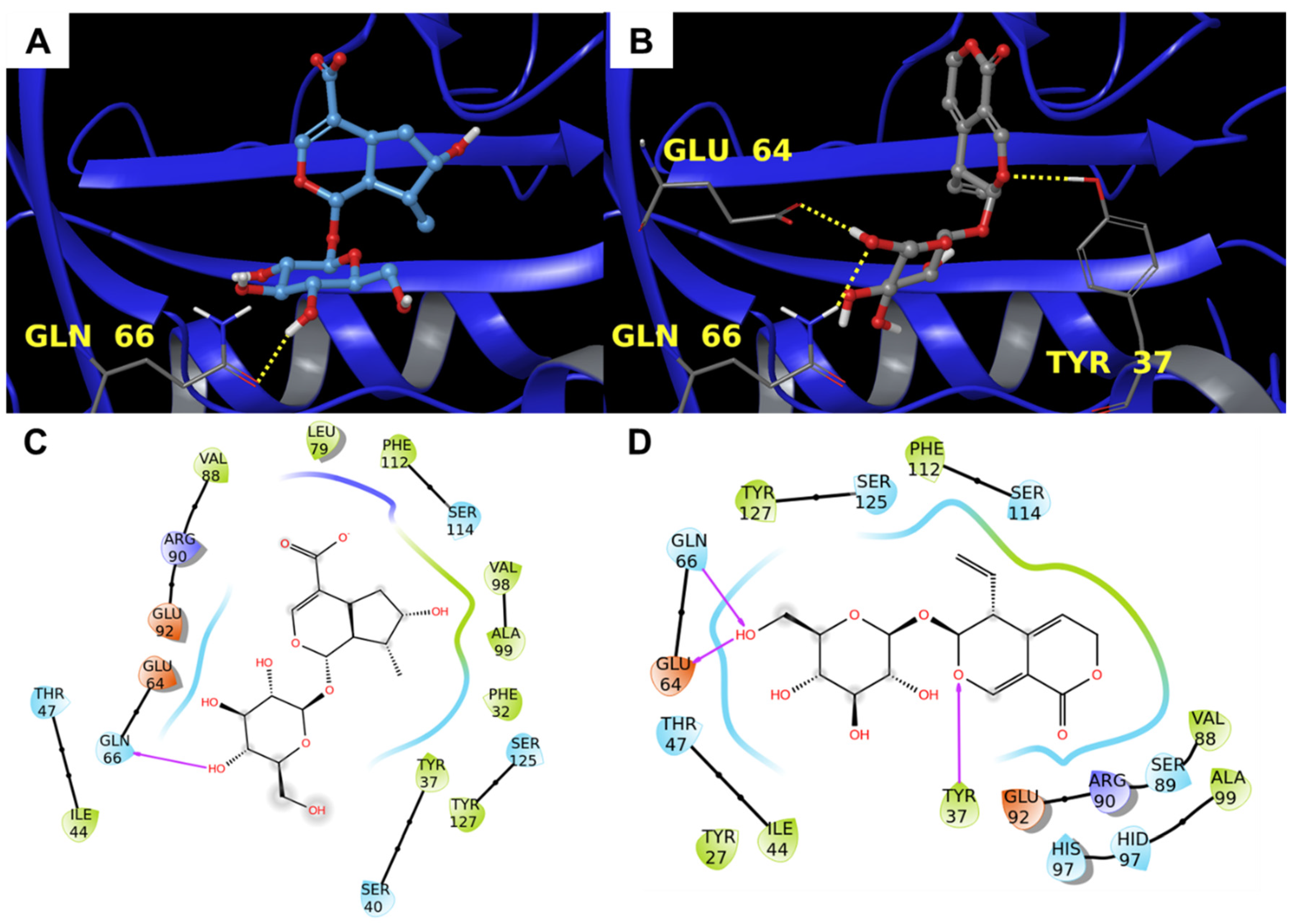
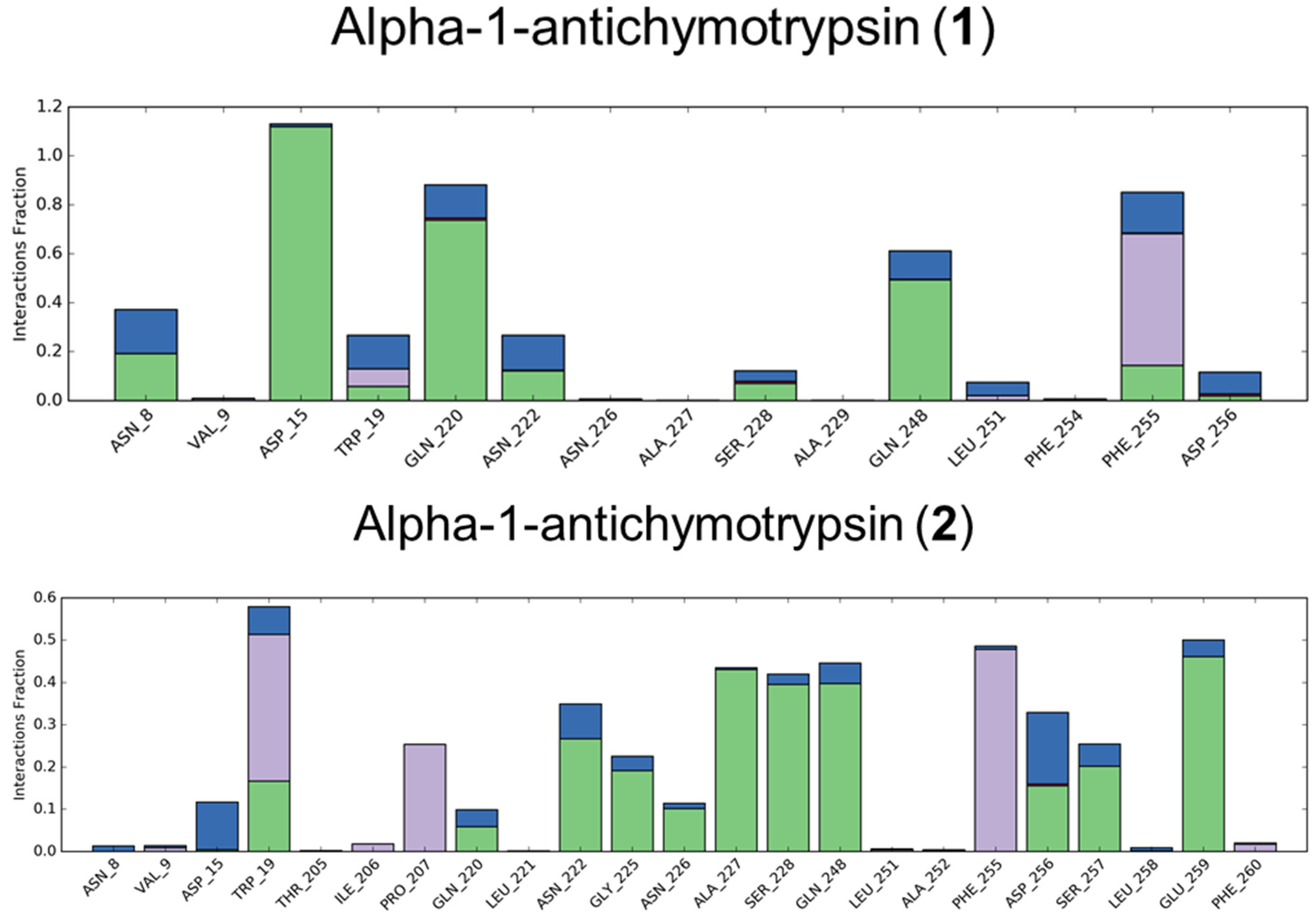
3.1.1. Alpha-1-antichymotrypsin
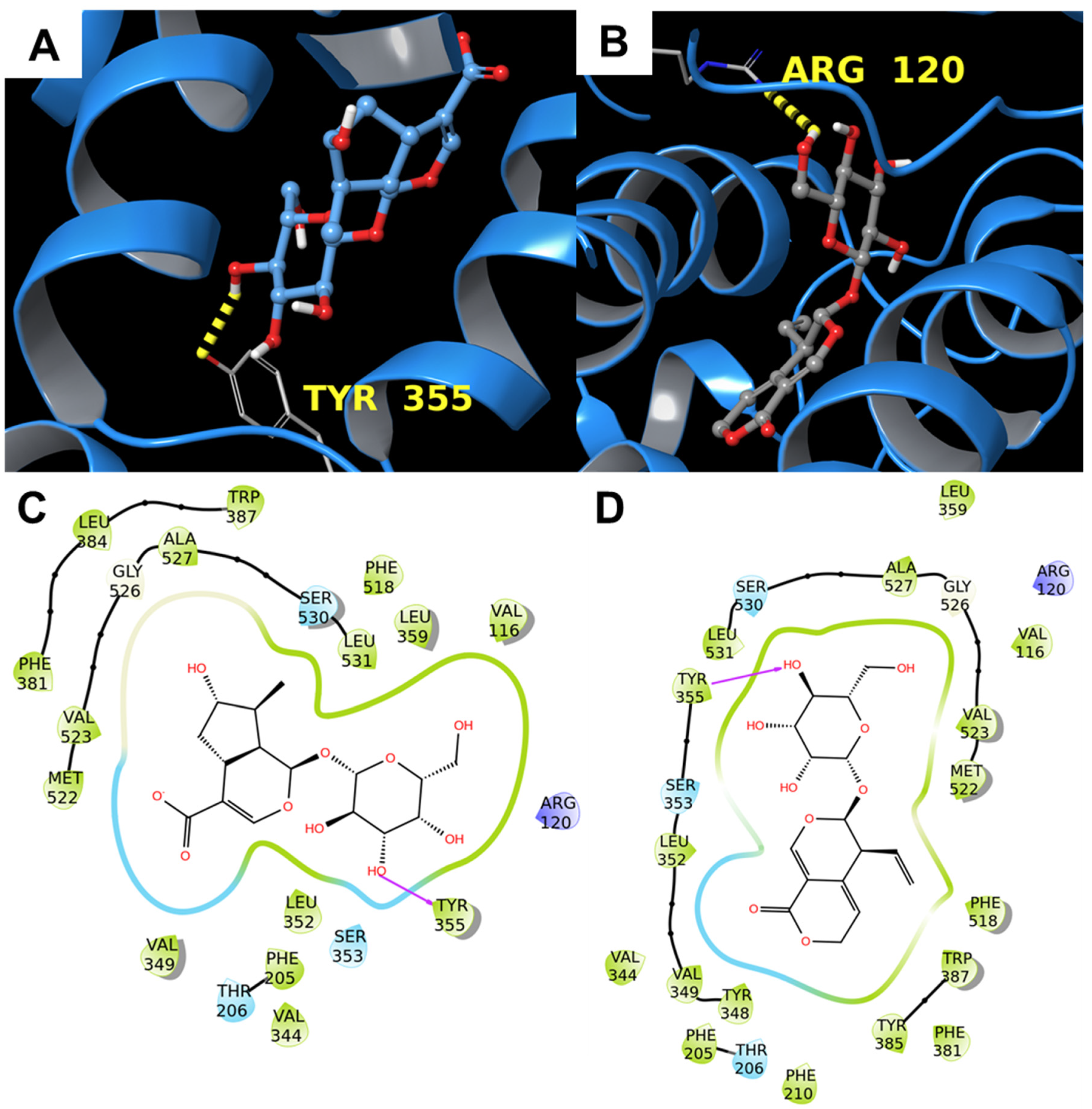
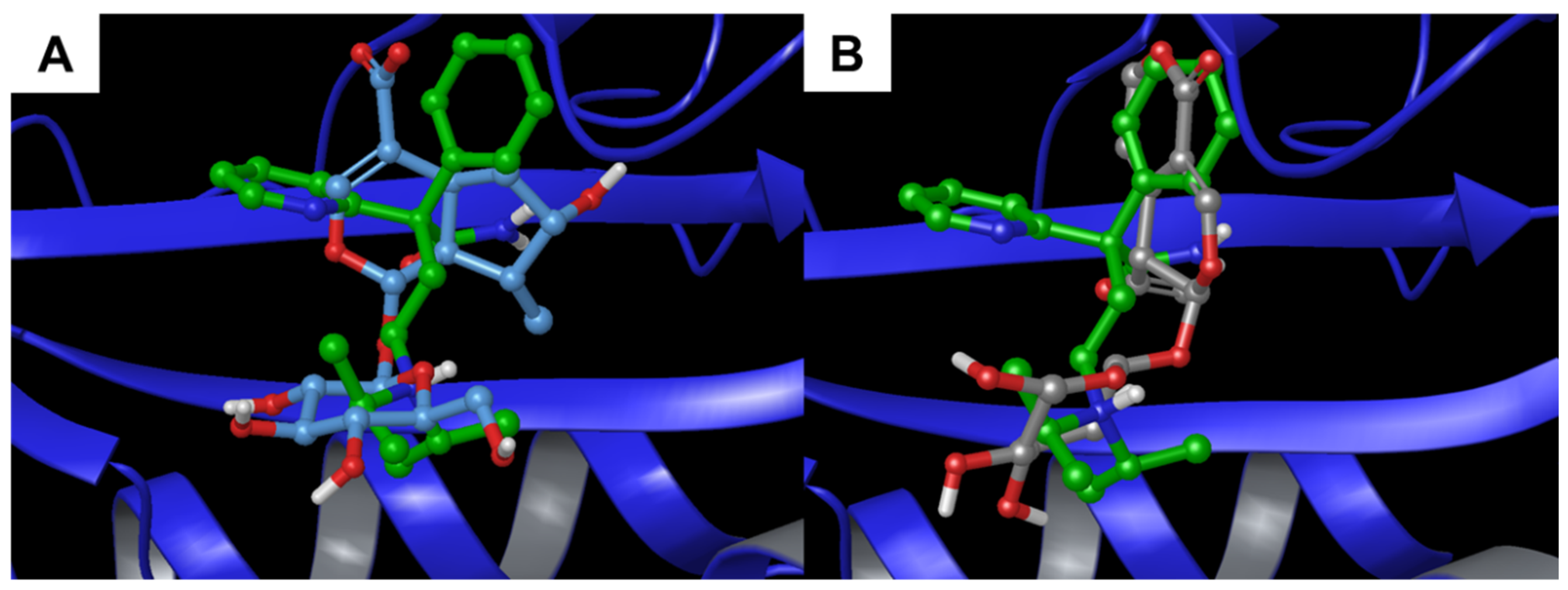
3.1.2. COX-2
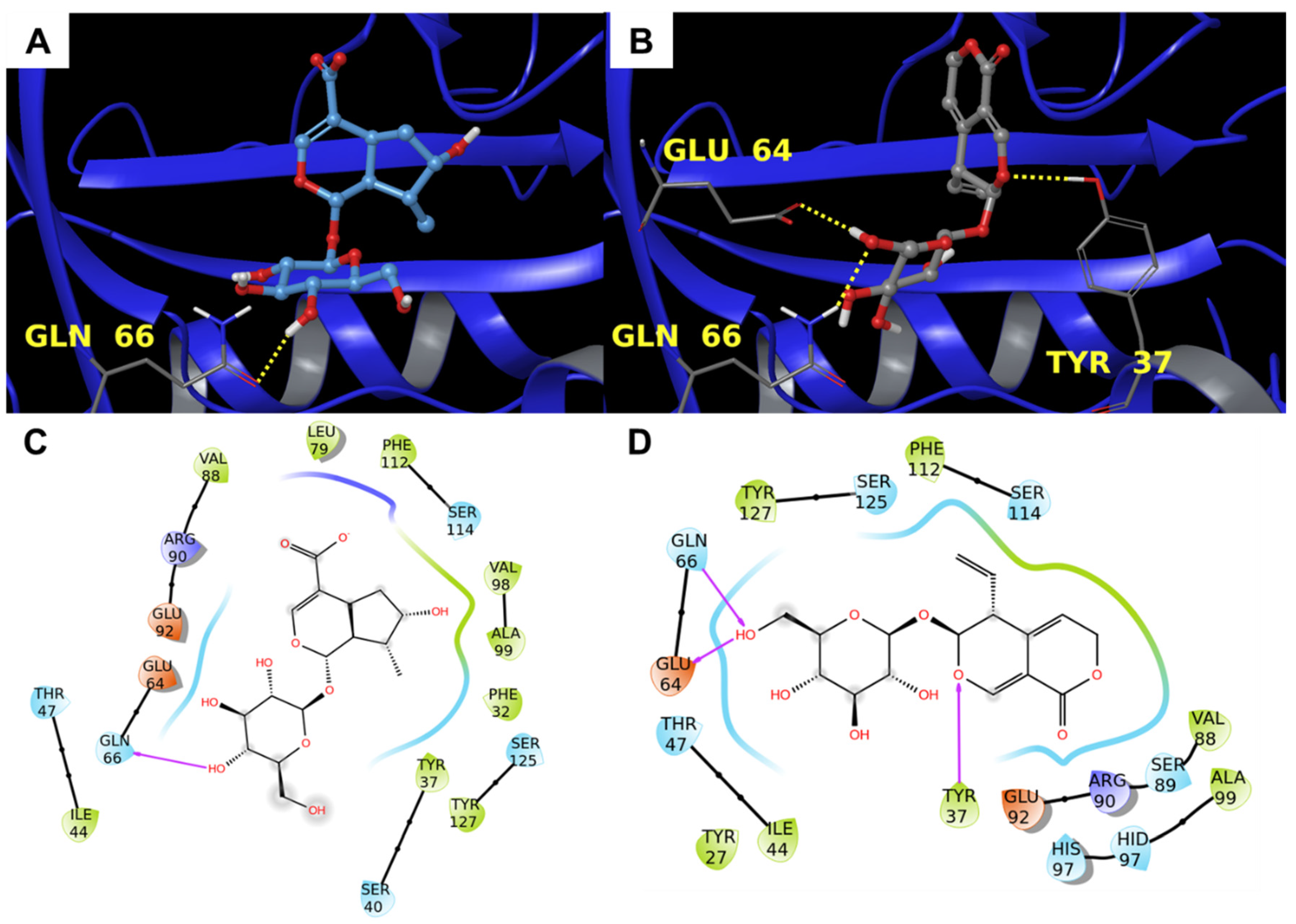
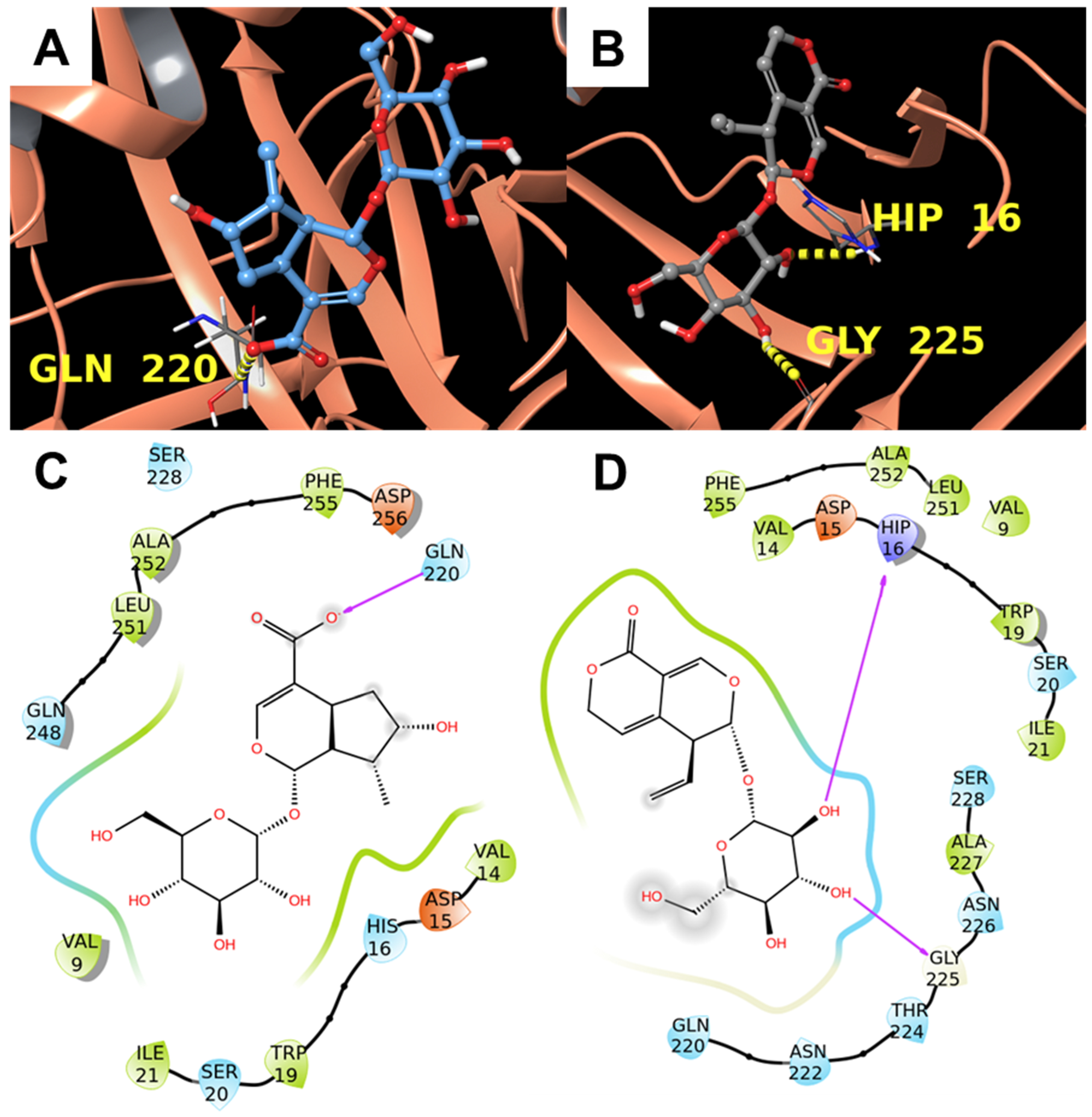
3.1.3. Alpha-1-Acid Glycoprotein
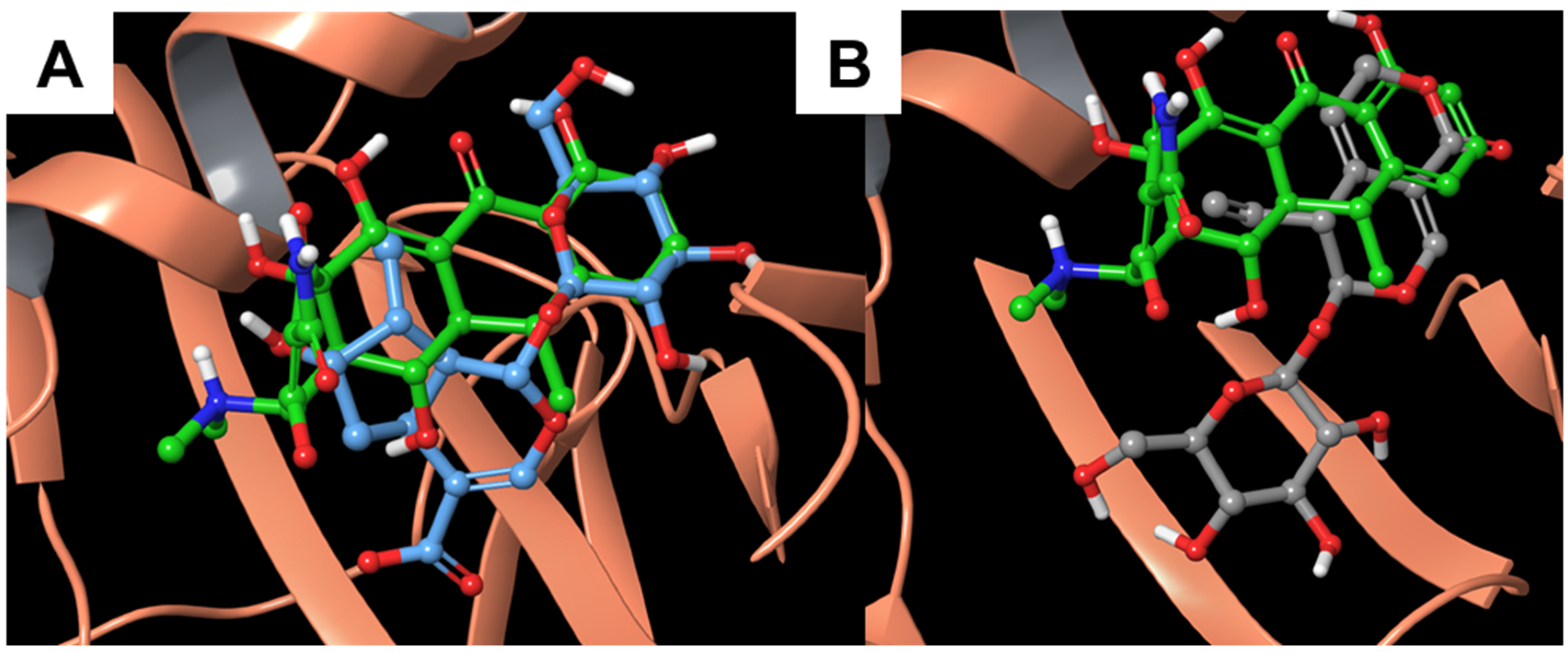
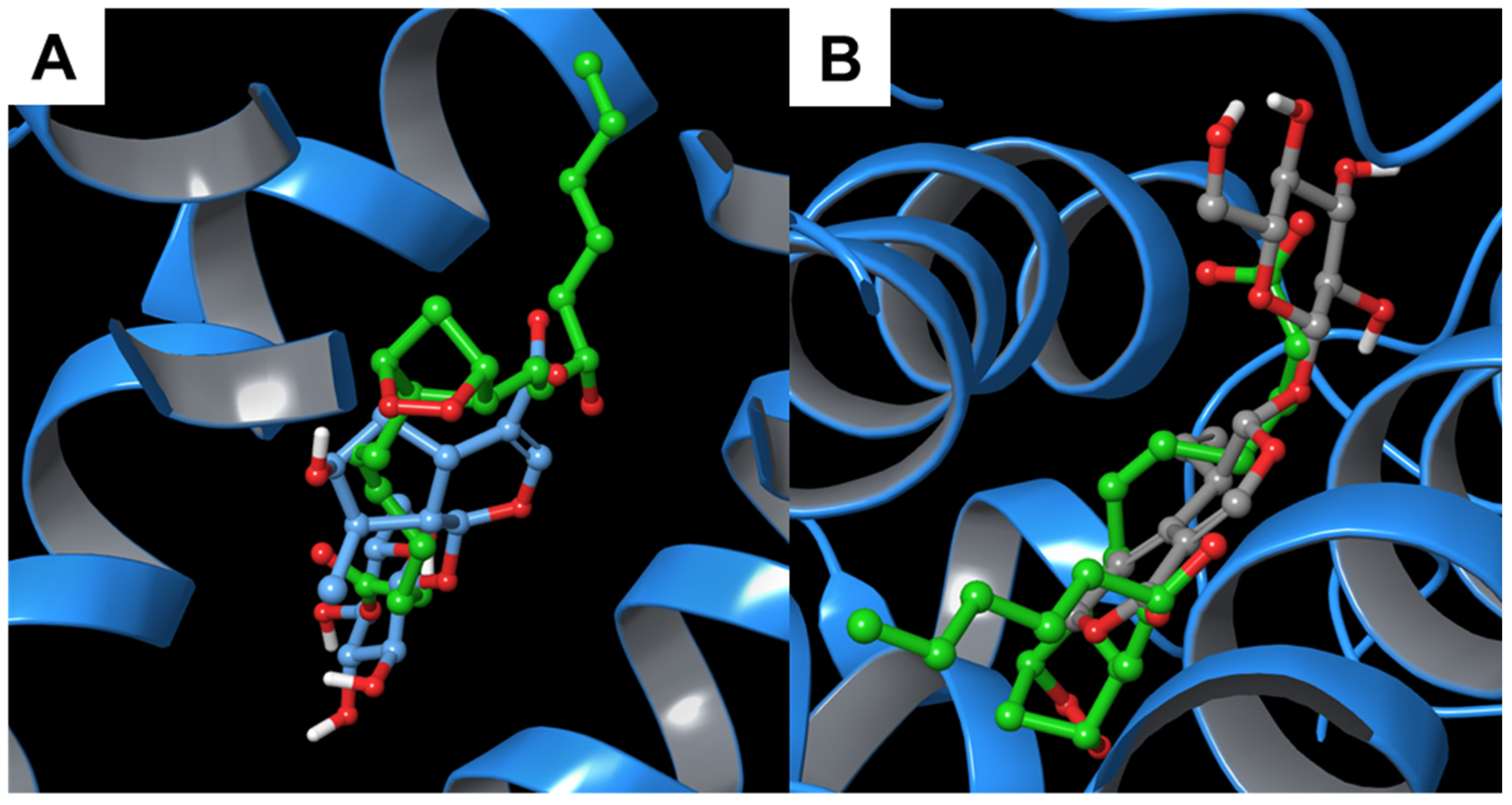
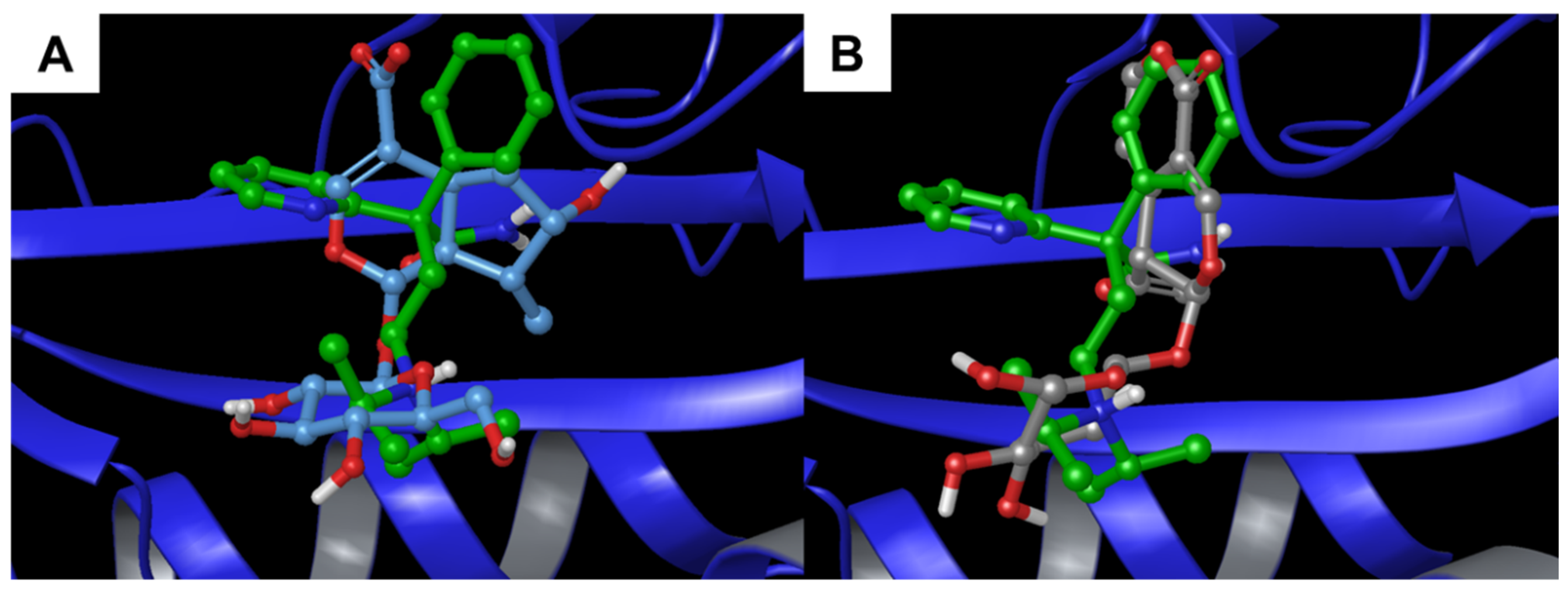
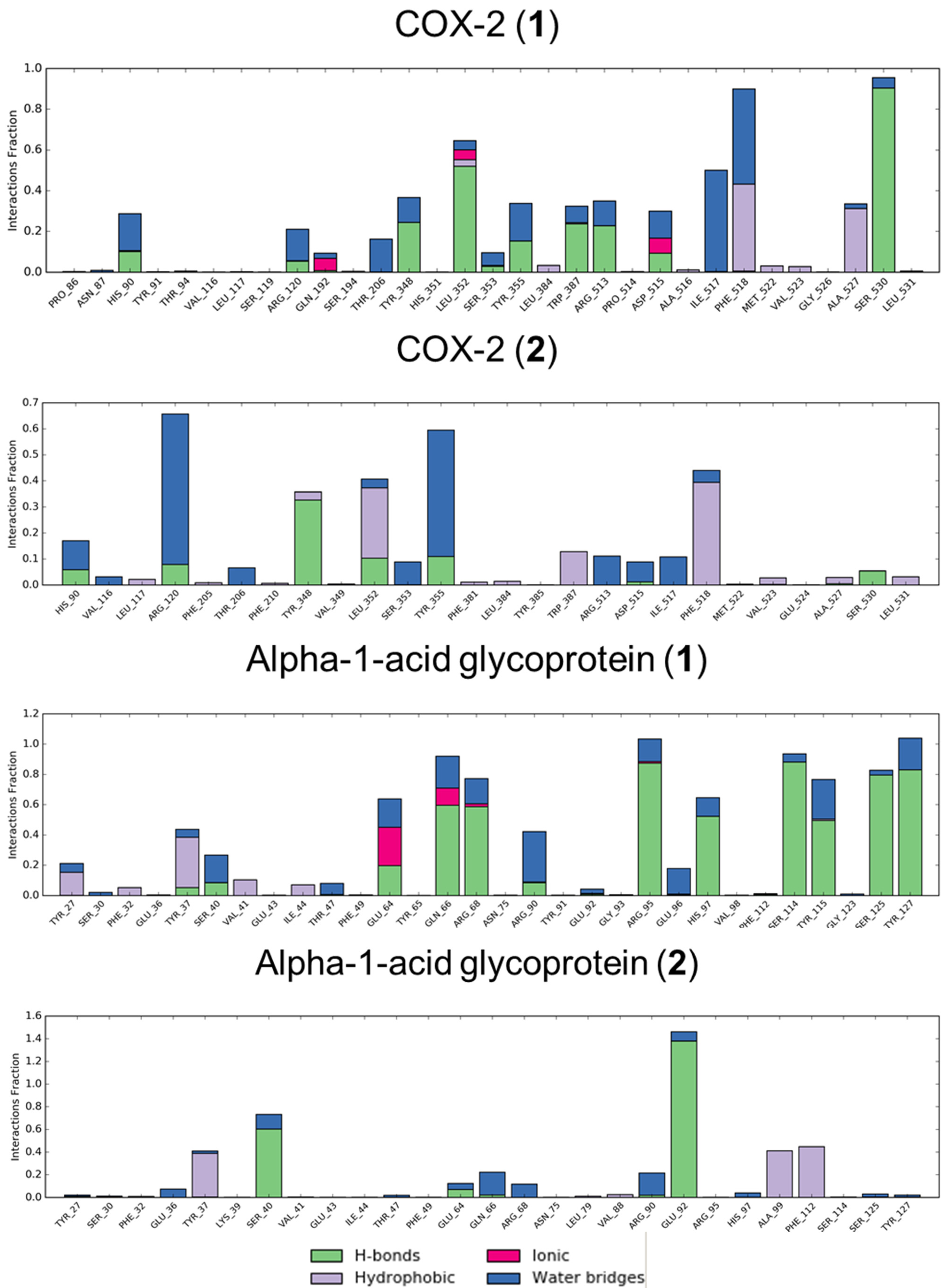
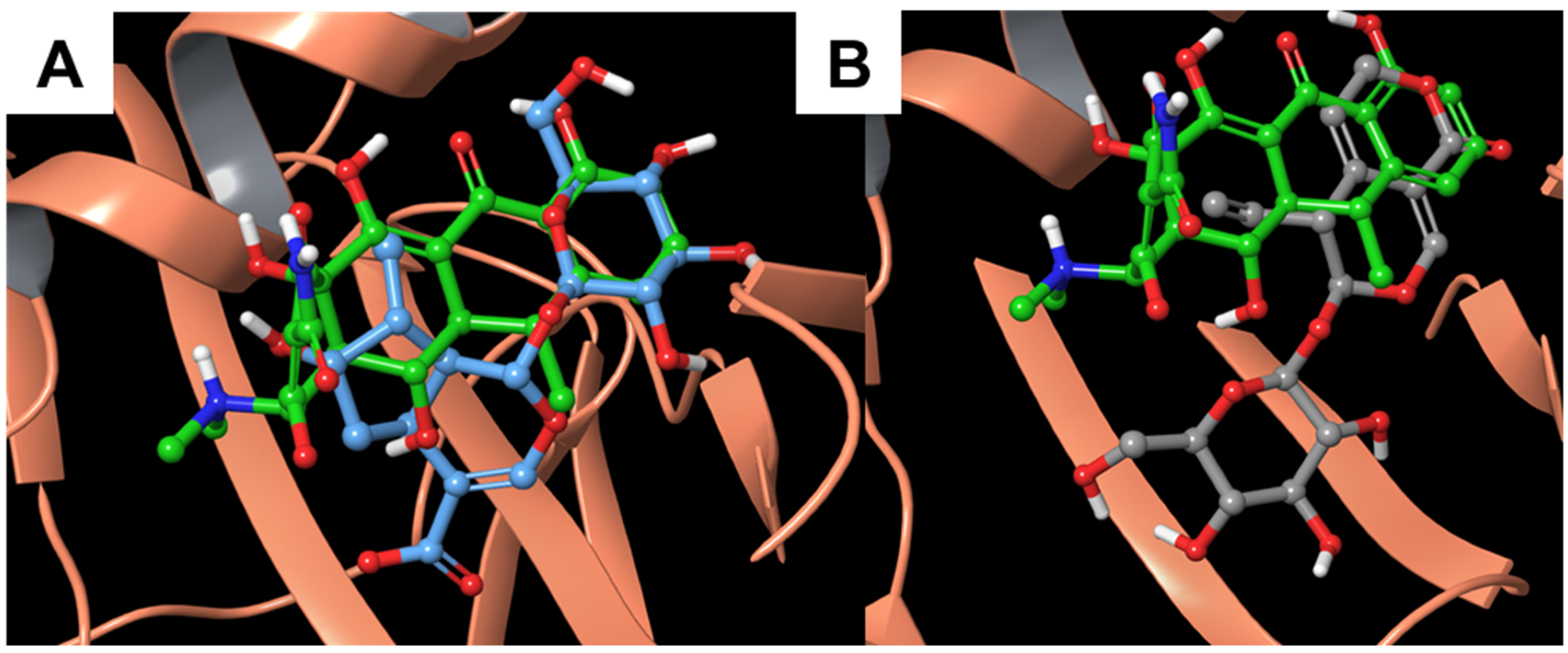
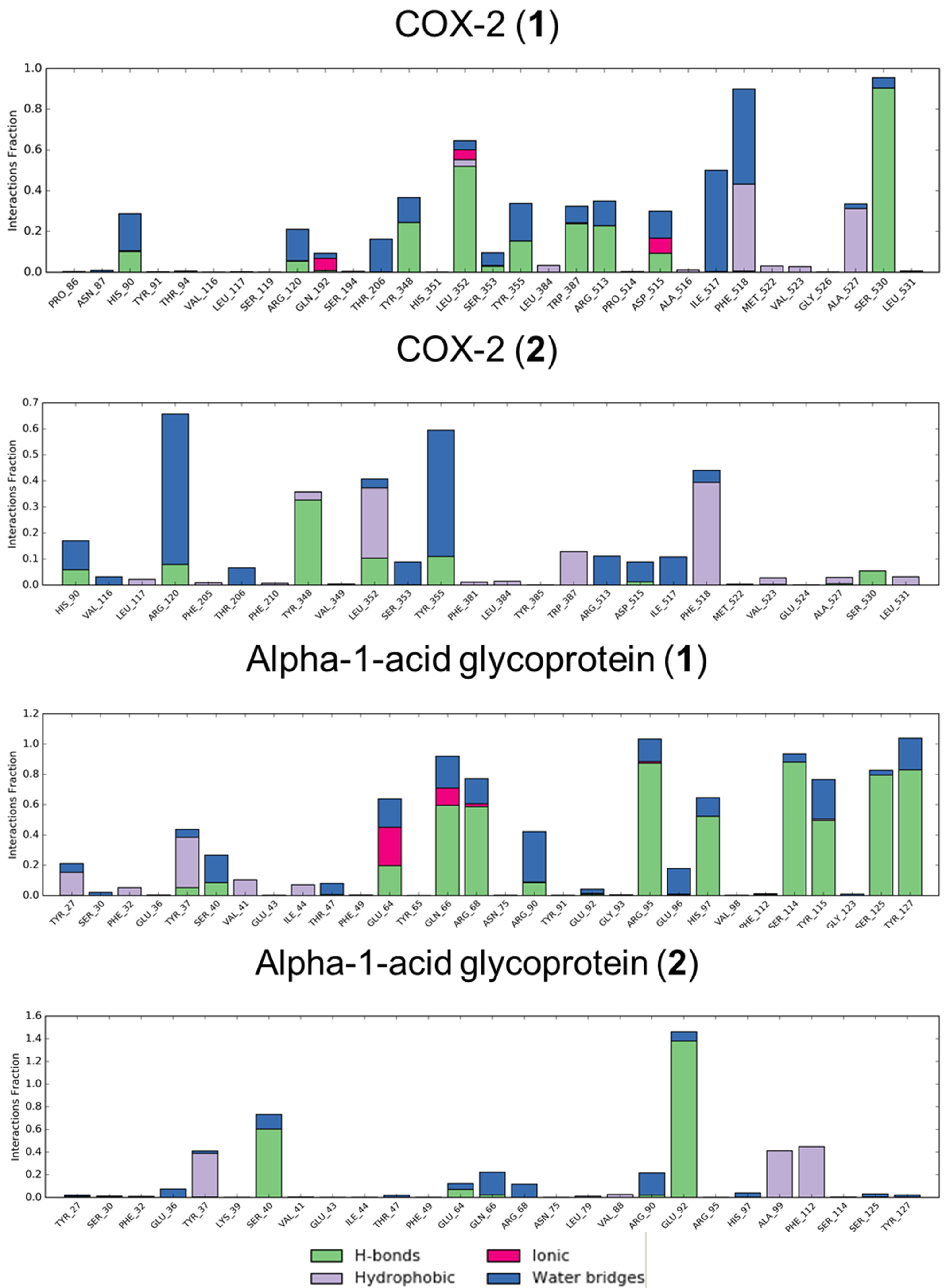
3.2. Molecular Dynamics
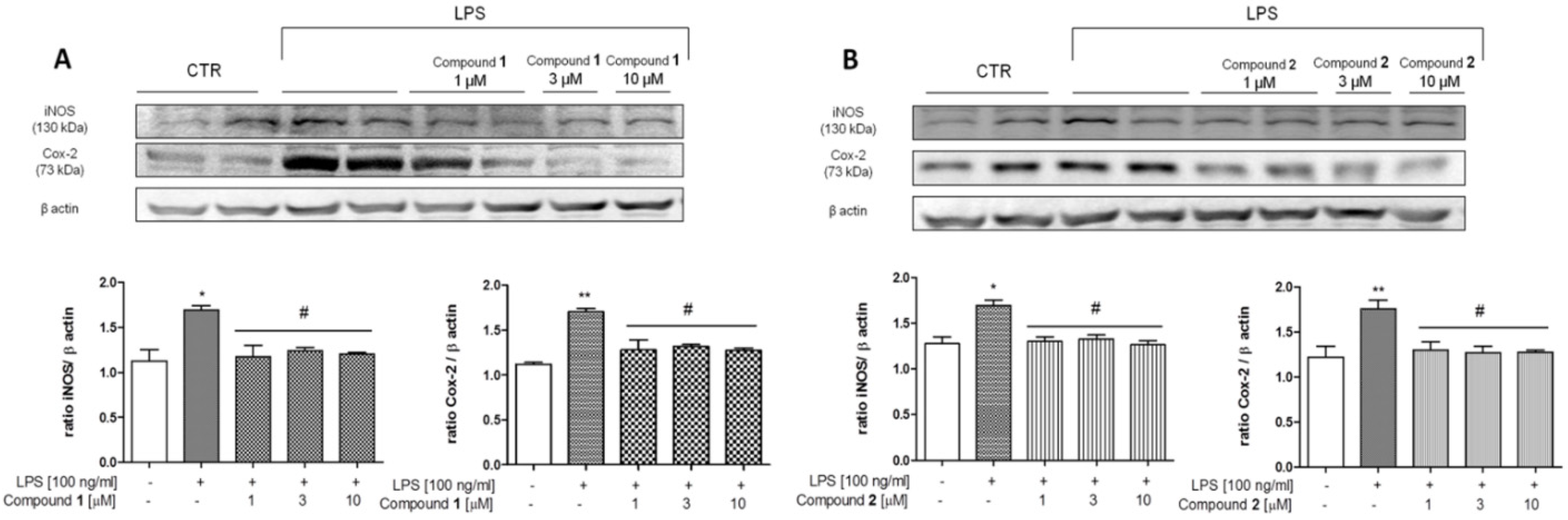
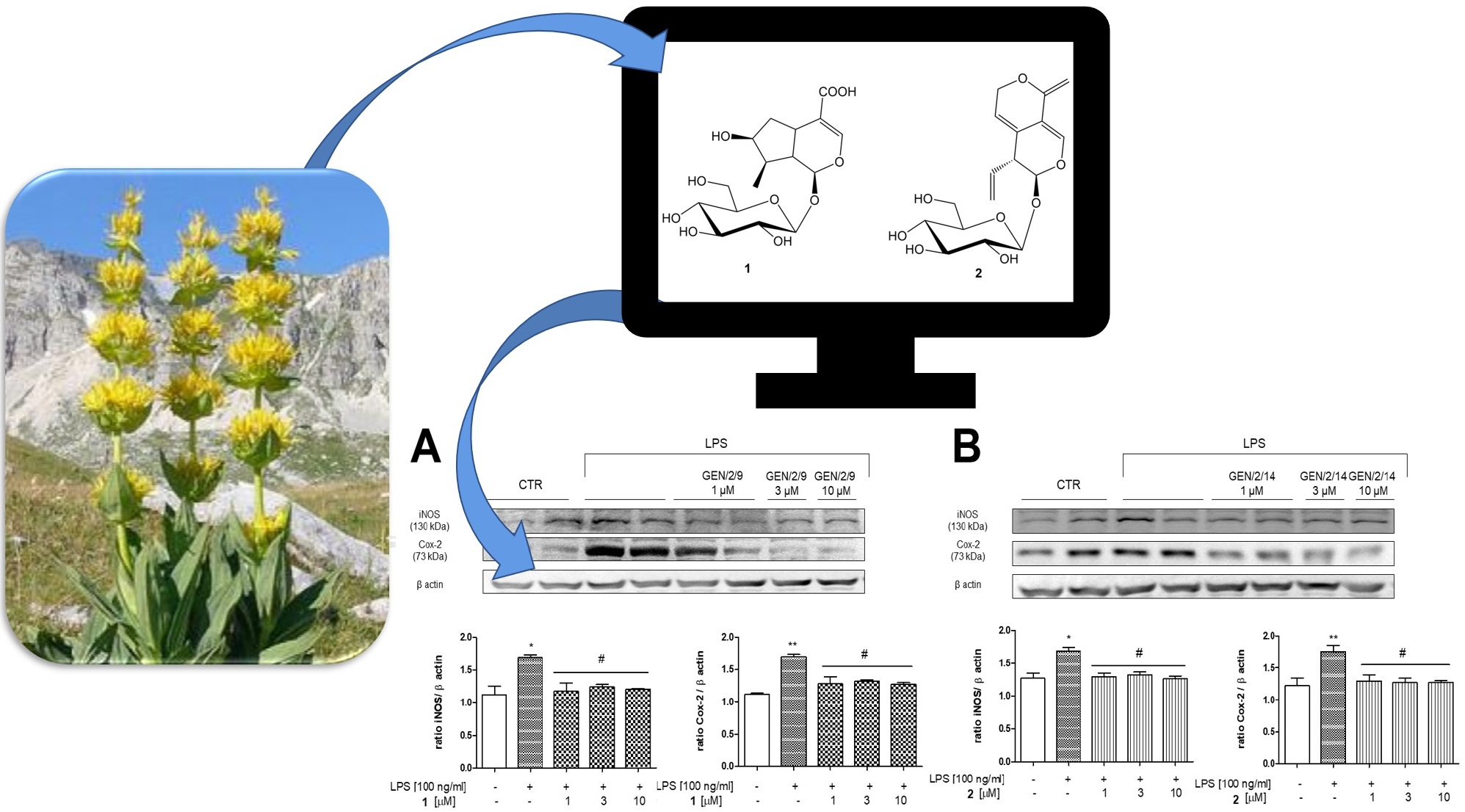
3.3. In Vitro Analysis
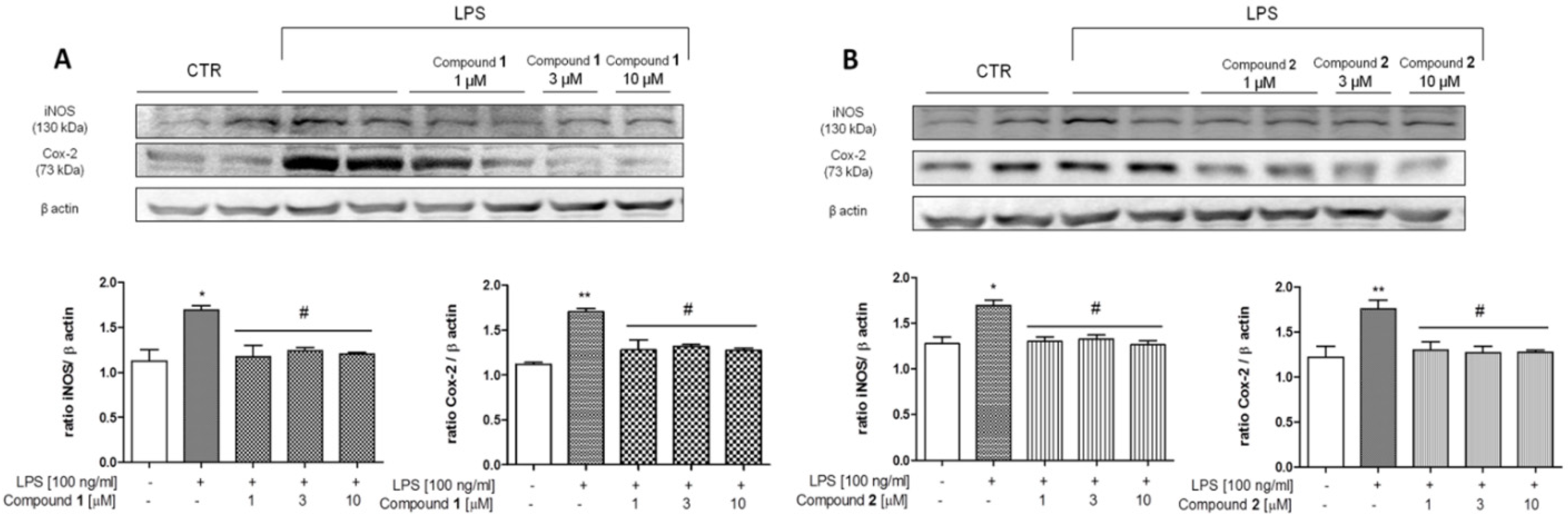
Effect of 1 and 2 on COX-2 and iNOS Expression in J774A.1 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Zhao, Y.-L.; Zhang, J.; Li, W.-Y.; Wang, Y.-Z. Phytochemistry and pharmacological activities of the genus Gentiana (Gentianaceae). Chem. Biodivers. 2016, 13, 107–150. [Google Scholar] [CrossRef] [PubMed]
- Cafaro, T.; Carnicelli, V.; Caprioli, G.; Maggi, F.; Celenza, G.; Perilli, M.; Bozzi, A.; Amicosante, G.; Brisdelli, F. Anti-apoptotic and anti-inflammatory activity of Gentiana lutea root extract. Adv. Trad. Med. 2020, 20, 619–630. [Google Scholar] [CrossRef]
- Kim, K.S.; Han, C.Y.; Han, Y.T.; Bae, E.J. Rhodanthpyrone A and B play an anti-inflammatory role by suppressing the nuclear factor-κB pathway in macrophages. Korean J. Physiol. Pharmacol. 2019, 23, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.-T.; Zhao, Y.-P.; Jiao, Y.-F.; Zheng, J.; Yang, W.-L.; Zhou, R.; Niu, Y.; Sun, T.; Li, Y.-X.; Yu, J.-Q. Protective effect of gentiopicroside against dextran sodium sulfate induced colitis in mice. Int. Immunopharmacol. 2016, 39, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, R.; Potunuru, U.R.; Nastasijević, B.T.A.; Joksić, G.; Dixit, M. Inhibition of vascular smooth muscle cell proliferation by Gentiana lutea root extracts. PLoS ONE 2013, 8, e61393. [Google Scholar] [CrossRef] [Green Version]
- Prakash, O.; Singh, R.; Kumar, S.; Srivastava, S.; Ved, A. Gentiana lutea Linn.(Yellow Gentian): A comprehensive. J. Ayu. Herb. Med. 2017, 3, 175–181. [Google Scholar]
- Xu, Y.; Li, Y.; Maffucci, K.G.; Huang, L.; Zeng, R. Analytical methods of phytochemicals from the genus Gentiana. Molecules 2017, 22, 2080. [Google Scholar] [CrossRef] [Green Version]
- Hudecová, A.; Hašplová, K.; Miadoková, E.; Magdolenová, Z.; Rinna, A.; Collins, A.R.; Gálová, E.; Vaculčíková, D.; Gregáň, F.; Dušinská, M. Gentiana asclepiadea protects human cells against oxidation DNA lesions. Cell Biochem. Funct. 2012, 30, 101–107. [Google Scholar] [CrossRef]
- Ando, H.; Hirai, Y.; Fujii, M.; Hori, Y.; Fukumura, M.; Niiho, Y.; Nakajima, Y.; Shibata, T.; Toriizuka, K.; Ida, Y. The chemical constituents of fresh Gentian Root. J. Nat. Med. 2007, 61, 269–279. [Google Scholar] [CrossRef]
- Mustafa, A.M.; Ricciutelli, M.; Maggi, F.; Sagratini, G.; Vittori, S.; Caprioli, G. Simultaneous determination of 18 bioactive compounds in Italian bitter liqueurs by reversed-phase high-performance liquid chromatography–diode array detection. Food Anal. Methods 2014, 7, 697–705. [Google Scholar] [CrossRef]
- Šavikin, K.; Menković, N.; Zdunić, G.; Stević, T.; Radanović, D.; Janković, T. Antimicrobial activity of Gentiana lutea L. extracts. Z. Naturforsch. C 2009, 64, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Menkovic, N.; Savikin-Fodulovic, K.; Cebedzic, R. Investigation of the activity of Gentiana lutea extracts against Mycobacterium bovis. Pharm. Pharmacol. Lett. 1999, 9, 74–75. [Google Scholar]
- Mathew, A.; Taranalli, A.D.; Torgal, S.S. Evaluation of anti-inflammatory and wound healing activity of Gentiana lutea rhizome extracts in animals. Pharm. Biol. 2004, 42, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Niiho, Y.; Yamazaki, T.; Nakajima, Y.; Yamamoto, T.; Ando, H.; Hirai, Y.; Toriizuka, K.; Ida, Y. Gastroprotective effects of bitter principles isolated from Gentian root and Swertia herb on experimentally-induced gastric lesions in rats. J. Nat. Med. 2006, 60, 82–88. [Google Scholar] [CrossRef]
- Mustafa, A.M.; Caprioli, G.; Dikmen, M.; Kaya, E.; Maggi, F.; Sagratini, G.; Vittori, S.; Öztürk, Y. Evaluation of neuritogenic activity of cultivated, wild and commercial roots of Gentiana lutea L. J. Funct. Foods 2015, 19, 164–173. [Google Scholar] [CrossRef]
- Patenković, A.; Stamenković-Radak, M.; Nikolić, D.; Marković, T.; Anđelković, M. Synergistic effect of Gentiana lutea L. on methyl methanesulfonate genotoxicity in the Drosophila wing spot test. J. Ethnopharmacol. 2013, 146, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Lauro, G.; Masullo, M.; Piacente, S.; Riccio, R.; Bifulco, G. Inverse Virtual Screening allows the discovery of the biological activity of natural compounds. Biorg. Med. Chem. 2012, 20, 3596–3602. [Google Scholar] [CrossRef]
- Lauro, G.; Romano, A.; Riccio, R.; Bifulco, G. Inverse Virtual Screening of antitumor targets: Pilot study on a small database of natural bioactive compounds. J. Nat. Prod. 2011, 74, 1401–1407. [Google Scholar] [CrossRef]
- Chini, M.G.; Lauro, G.; Bifulco, G. Addressing the target identification and accelerating the repositioning of anti-inflammatory/anti-cancer organic compounds by computational approaches. Eur. J. Org. Chem. 2021, 2021, 2966–2981. [Google Scholar] [CrossRef]
- De Marino, S.; Festa, C.; Zollo, F.; Iorizzi, M. Novel steroidal components from the underground parts of Ruscus aculeatus L. Molecules 2012, 17, 14002–14014. [Google Scholar] [CrossRef] [Green Version]
- Mpondo Mpondo, E.; Chulia, A.J. 6′-O-β-D-Glucosyl Gentiopicroside: A New Secoiridoid from Gentiana asclepiadea. Planta Med. 1988, 54, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Calis, I.; Lahloub, M.F.; Sticher, O. Loganin, loganic acid and periclymenoside, a new biosidic ester iridoid glucoside from Lonicera periclymenum L. (Caprifoliaceae). Helv. Chim. Acta 1984, 67, 160–165. [Google Scholar] [CrossRef]
- De Vita, S.; Lauro, G.; Ruggiero, D.; Terracciano, S.; Riccio, R.; Bifulco, G. Protein preparation automatic protocol for high-throughput inverse virtual screening: Accelerating the target identification by computational methods. J. Chem. Inf. Model. 2019, 59, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-1: Maestro; Schrödinger LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-1: LigPrep; Schrödinger LLC: New York, NY, USA, 2017.
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 3–33. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2017-1: Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2017.
- Halgren, T.A. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2017. In Maestro-Desmond Interoperability Tools; Schrödinger, LLC: New York, NY, USA, 2020.
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Bakhle, Y.S. COX-2 and cancer: A new approach to an old problem. Br. J. Pharmacol. 2001, 134, 1137–1150. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Jaberie, H.; Hosseini, S.V.; Naghibalhossaini, F. Evaluation of alpha 1-antitrypsin for the early diagnosis of colorectal cancer. Pathol. Oncol. Res. 2020, 26, 1165–1173. [Google Scholar] [CrossRef]
- Jackson, P.R.; Tucker, G.T.; Woods, H.F. Altered plasma drug binding in cancer: Role of α1-acid glycoprotein and albumin. Clin. Pharmacol. Ther. 1982, 32, 295–302. [Google Scholar] [CrossRef]
- Torres-Durán, M.; Ruano-Ravina, A.; Parente-Lamelas, I.; Abal-Arca, J.; Leiro-Fernández, V.; Montero-Martínez, C.; Pena, C.; Castro-Añón, O.; Golpe-Gómez, A.; González-Barcala, F.J.; et al. Alpha-1 antitrypsin deficiency and lung cancer risk: A case-control study in never-smokers. J. Thorac. Oncol. 2015, 10, 1279–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Andersson, R.; Hu, D.; Bauden, M.; Sasor, A.; Bygott, T.; Pawłowski, K.; Pla_Parada, I.; Marko-Varga, G.; Ansari, D. Alpha-1-acid glycoprotein 1 (AGP1) as a novel biomarker for pancreatic cancer. J. Clin. Oncol. 2019, 37, e15708. [Google Scholar] [CrossRef]
- Gettins, P.G.W. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Gardill, B.R.; Kern, A.; Kirchweger, P.; Börsch, M.; Muller, Y.A. Design of an allosterically modulated doxycycline and doxorubicin drug-binding protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labib, M.B.; Sharkawi, S.M.Z.; El-Daly, M. Design, synthesis of novel isoindoline hybrids as COX-2 inhibitors: Anti-inflammatory, analgesic activities and docking study. Bioorg. Chem. 2018, 80, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Ermondi, G.; Caron, G.; Lawrence, R.; Longo, D. Docking studies on NSAID/COX-2 isozyme complexes using Contact Statistics analysis. J. Comput.-Aided Mol. Des. 2004, 18, 683–696. [Google Scholar] [CrossRef]
- Kiefer, J.R.; Pawlitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Gierse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef]
- Fournier, T.; Medjoubi-N, N.; Porquet, D. Alpha-1-acid glycoprotein. Biochim. Biophys. Acta 2000, 1482, 157–171. [Google Scholar] [CrossRef]
- Hochepied, T.; Berger, F.G.; Baumann, H.; Libert, C. α1-Acid glycoprotein: An acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor Rev. 2003, 14, 25–34. [Google Scholar] [CrossRef]
- Nishi, K.; Ono, T.; Nakamura, T.; Fukunaga, N.; Izumi, M.; Watanabe, H.; Suenaga, A.; Maruyama, T.; Yamagata, Y.; Curry, S.; et al. Structural insights into differences in drug-binding selectivity between two forms of human α1-acid glycoprotein genetic variants, the A and F1*S Forms. J. Biol. Chem. 2011, 286, 14427–14434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakar, K.A.; Lam, S.D.; Sidek, H.M.; Feroz, S.R. Characterization of the interaction of diosgenin with human serum albumin and α1-acid glycoprotein using biophysical and bioinformatic tools. J. Mol. Liq. 2020, 306, 112865. [Google Scholar] [CrossRef]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A novel mechanism of cyclooxygenase-2 inhibition Involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | PDB | V * | Calculated Binding Affinity (V0) | Average Decoy Binding Affinity (VR) | % of Occurrences |
|---|---|---|---|---|---|
| Transient Receptor Potential Cation Channel Subfamily V Member 1 | 5irz | 1.48 | −7.7 | −5.2 | 0.3% |
| Alpha-1-Acid Glycoprotein 2 | 3apw | 1.18 | −8.3 | −7.0 | 0.1% |
| Transient Receptor Potential Cation Channel Subfamily V Member 1 | 5is0 | 1.18 | −8.7 | −7.3 | 0.3% |
| Tumor Necrosis Factor Ligand Superfamily Member 4 | 2hew | 1.13 | −9.4 | −8.3 | 0.2% |
| Complement C3 Membrane Cofactor Protein | 5fo8 | 1.12 | −9.5 | −8.5 | 2.6% |
| Alpha-1-Antichymotrypsin | 5om2 | 1.12 | −8.6 | −7.7 | 1.2% |
| Tumor Necrosis Factor Ligand Superfamily Member 4 | 2hey | 1.11 | −9.2 | −8.2 | 0.2% |
| Alpha-1-Acid Glycoprotein 2 | 3apu | 1.10 | −9.1 | −8.3 | 0.1% |
| Prothrombin | 4ax9 | 1.10 | −7.8 | −7.1 | 23.3% |
| Prostaglandin H2 Synthase-2 | 1ddx | 1.09 | −9.5 | −8.7 | 0.6% |
| Target | PDB | V * | Calculated Binding Affinity (V0) | Average Decoy Binding Affinity (VR) | % of Occurrences |
|---|---|---|---|---|---|
| Alpha-1-Acid Glycoprotein 2 | 3apw | 1.21 | −8.5 | −7.0 | 0.1% |
| Alpha-1-Antichymotrypsin | 5om2 | 1.13 | −8.7 | −7.7 | 1.2% |
| Alpha-1-Antichymotrypsin | 5om3 | 1.10 | −8.6 | −7.8 | 1.2% |
| Prostaglandin H2 Synthase-2 | 1ddx | 1.09 | −9.5 | −8.7 | 1.0% |
| Tumor Necrosis Factor Ligand Superfamily member 4 | 2hew | 1.09 | −9.0 | −8.3 | 0.1% |
| Alpha-1-Antichymotrypsin | 5om7 | 1.08 | −8.3 | −7.7 | 1.2% |
| Tyrosine-Protein Kinase BTK | 6aub | 1.07 | −8.0 | −7.4 | 3.2% |
| Alpha-1-Antichymotrypsin | 5om7 | 1.06 | −8.3 | −7.8 | 1.2% |
| Tyrosine-Protein Kinase BTK | 6bik | 1.06 | -8.1 | −7.6 | 3.2% |
| Tumor Necrosis Factor Ligand Superfamily Member 4 | 2hey | 1.05 | −8.7 | −8.28 | 0.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vita, S.; Chini, M.G.; Saviano, G.; Finamore, C.; Festa, C.; Lauro, G.; De Marino, S.; Russo, R.; Avagliano, C.; Casapullo, A.; et al. Biological Profile of Two Gentiana lutea L. Metabolites Using Computational Approaches and In Vitro Tests. Biomolecules 2021, 11, 1490. https://doi.org/10.3390/biom11101490
De Vita S, Chini MG, Saviano G, Finamore C, Festa C, Lauro G, De Marino S, Russo R, Avagliano C, Casapullo A, et al. Biological Profile of Two Gentiana lutea L. Metabolites Using Computational Approaches and In Vitro Tests. Biomolecules. 2021; 11(10):1490. https://doi.org/10.3390/biom11101490
Chicago/Turabian StyleDe Vita, Simona, Maria Giovanna Chini, Gabriella Saviano, Claudia Finamore, Carmen Festa, Gianluigi Lauro, Simona De Marino, Roberto Russo, Carmen Avagliano, Agostino Casapullo, and et al. 2021. "Biological Profile of Two Gentiana lutea L. Metabolites Using Computational Approaches and In Vitro Tests" Biomolecules 11, no. 10: 1490. https://doi.org/10.3390/biom11101490
APA StyleDe Vita, S., Chini, M. G., Saviano, G., Finamore, C., Festa, C., Lauro, G., De Marino, S., Russo, R., Avagliano, C., Casapullo, A., Calignano, A., Bifulco, G., & Iorizzi, M. (2021). Biological Profile of Two Gentiana lutea L. Metabolites Using Computational Approaches and In Vitro Tests. Biomolecules, 11(10), 1490. https://doi.org/10.3390/biom11101490