
Osteogenesis Imperfecta: Current and Prospective Therapies

 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Review
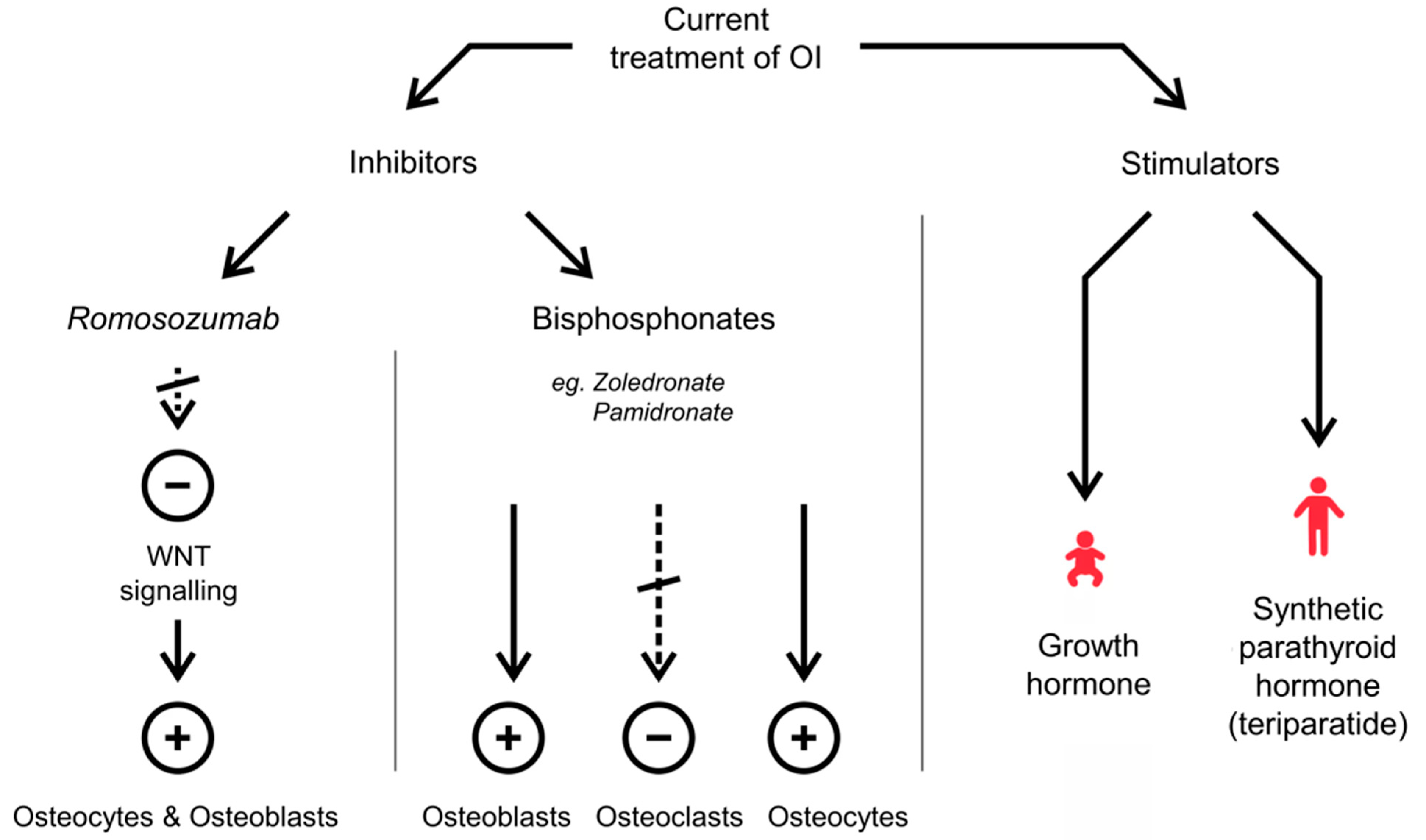
2.1. Current Treatment of OI
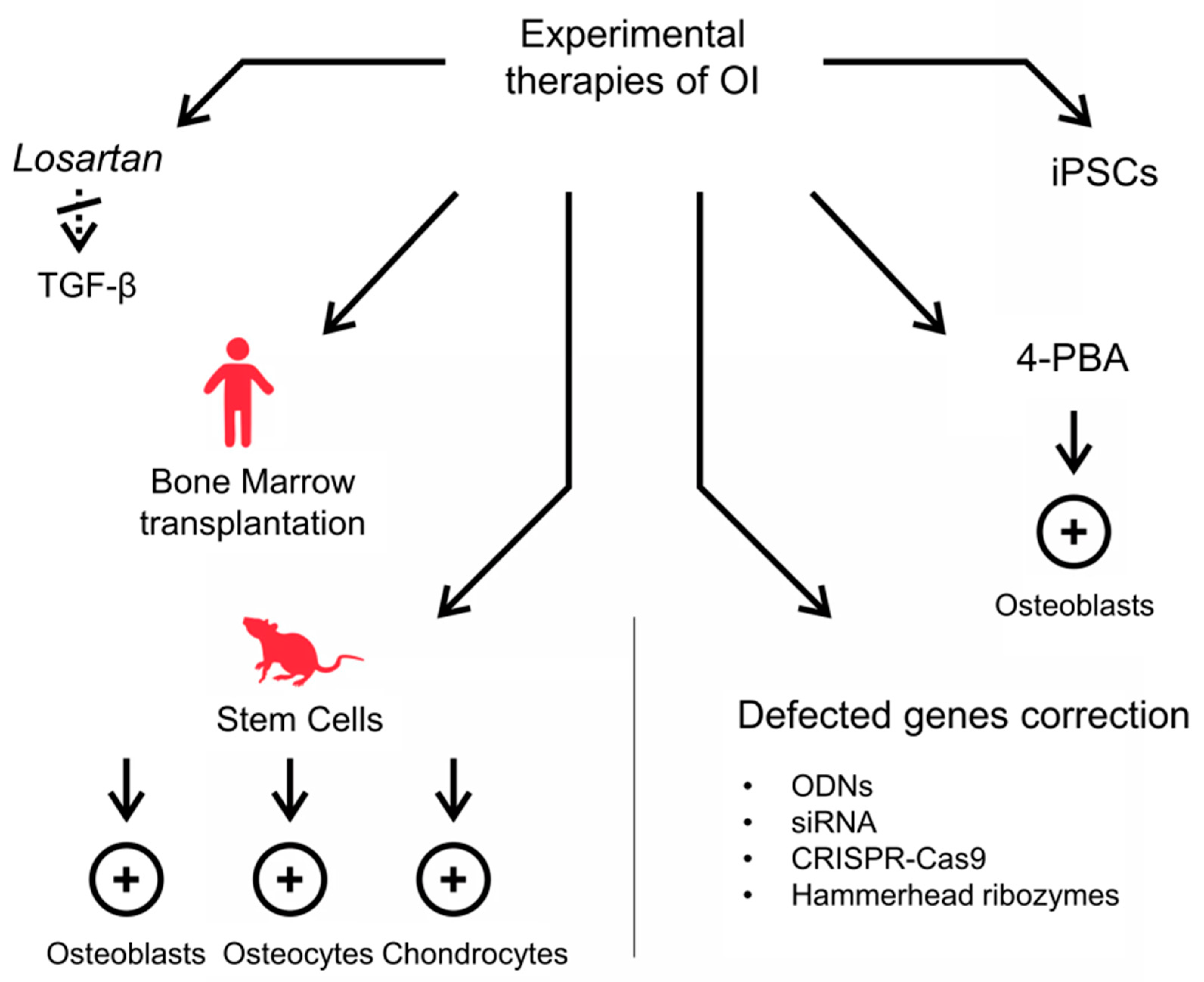
2.2. Experimental Strategies for OI Therapy
2.2.1. Anti-TGF-β Antibodies
2.2.2. Stem Cells Transplantation
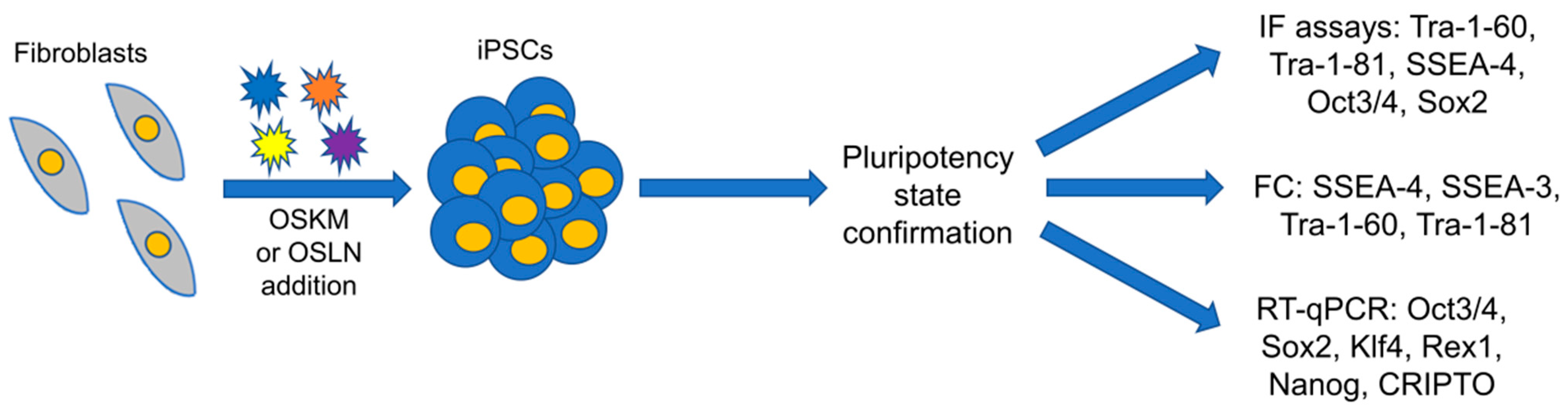
2.2.3. Methods Based on Genetic Engineering and Somatic Cells Reprogramming into iPSCs
2.2.4. Counteraction of ER Stress and UPR
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sillence, O.D.; Senn, A.; Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979, 16, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Chu, M.-L.; Williams, C.J.; Pepe, G.; Hirsch, J.L.; Prockop, D.J.; Ramirez, F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nat. Cell Biol. 1983, 304, 78–80. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part A 2019, 179, 2393–2419. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31633310 (accessed on 2 November 2019). [CrossRef] [PubMed]
- Landis, W.J.; Hodgens, K.J.; Song, M.J.; Arena, J.; Kiyonaga, S.; Marko, M.; Owen, C.; McEwen, B.F. Mineralization of collagen may occur on fibril surfaces: Evidence from conven-tional and high-voltage electron microscopy and three-dimensional imaging. J. Struct. Biol. 1996, 117, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Forlino, A.; Cabral, W.A.; Barnes, A.; Marini, J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011, 7, 540–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, A.; Chang, W.; Morello, R.; Cabral, W.A.; Weis, M.; Eyre, D.R.; Leikin, S.; Makareeva, E.; Kuznetsova, N.; Uveges, T.E.; et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006, 355, 2757–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006, 127, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, D.; Schwarze, U.; Morello, R.; Lennington, J.; Bertin, T.K.; Pace, J.M.; Pepin, M.G.; Weis, M.; Eyre, D.R.; Walsh, J.; et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008, 29, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Rauch, F.; Moffatt, P.; Cheung, M.; Roughley, P.; Lalic, L.; Lund, A.M.; Ramirez, N.; Fahiminiya, S.; Majewski, J.; Glorieux, F.H. Osteogenesis imperfecta type V: Marked phenotypic variability despite the presence of the IFITM5c.−14C > T mutation in all patients. J. Med. Genet. 2013, 50, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Homan, E.P.; Rauch, F.; Grafe, I.; Lietman, C.; Doll, A.J.; Dawson, B.; Bertin, T.; Napierala, D.; Morello, R.; Gibbs, R.; et al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J. Bone Miner. Res. 2011, 26, 2798–2803. [Google Scholar] [CrossRef]
- Christiansen, H.E.; Schwarze, U.; Pyott, S.M.; Alswaid, A.; Al Balwi, M.; Alrasheed, S.; Pepin, M.G.; Weis, M.A.; Eyre, D.R.; Byers, P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Barnes, A.; Cabral, W.A.; Weis, M.; Makareeva, E.; Mertz, E.L.; Leikin, S.; Eyre, D.; Trujillo, C.; Marini, J.C. Absence of FKBP10in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. 2012, 33, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollitt, R.C.; Saraff, V.; Dalton, A.; Webb, E.A.; Shaw, N.J.; Sobey, G.J.; Mughal, M.Z.; Hobson, E.; Ali, F.; Bishop, N.J.; et al. Phenotypic variability in patients with osteogenesis imperfecta caused by BMP1 muta-tions. Am. J. Med. Genet. Part A 2016, 170, 3150–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahiminiya, S.; Majewski, J.; Mort, J.; Moffatt, P.; Glorieux, F.H.; Rauch, F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J. Med. Genet. 2013, 50, 345–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapunzina, P.; Aglan, M.; Temtamy, S.; Caparrós-Martín, J.A.; Valencia, M.; Letón, R.; Martínez-Glez, V.; Elhossini, R.; Amr, K.; Vilaboa, N.; et al. Identification of a frameshift mutation in osterix in a patient with recessive osteo-genesis imperfecta. Am. J. Hum. Genet. 2010, 87, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Naka, H. Bisphosphonates and bone quality. Clin. Calcium 2005, 15, 1020–1025. [Google Scholar]
- Martin, T.J.; Seeman, E. Bone remodelling: Its local regulation and the emergence of bone fragility. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 701–722. [Google Scholar] [CrossRef]
- Nijhuis, W.H.; Eastwood, D.M.; Allgrove, J.; Hvid, I.; Weinans, H.H.; Bank, R.A.; Sakkers, R.J. Current concepts in osteogenesis imperfecta: Bone structure, biomechanics and medical management. J. Child. Orthop. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Nasomyont, N.; Hornung, L.; Wasserman, H. Intravenous bisphosphonate therapy in children with spinal muscular atrophy. Osteoporos. Int. 2019, 31, 995–1000. [Google Scholar] [CrossRef]
- Wasserman, H.M.; Hornung, L.N.; Stenger, P.J.; Rutter, M.M.; Wong, B.L.; Rybalsky, I.; Khoury, J.C.; Kalkwarf, H.J. Low bone mineral density and fractures are highly prevalent in pediatric patients with spinal muscular atrophy regardless of disease severity. Neuromuscul. Disord. 2017, 27, 331–337. [Google Scholar] [CrossRef]
- Borah, B.; Dufresne, T.; Nurre, J.; Phipps, R.; Chmielewski, P.; Wagner, L.; Lundy, M.; Bouxsein, M.; Zebaze, R.; Seeman, E. Risedronate reduces intracortical porosity in women with osteoporosis. J. Bone Miner. Res. 2009, 25, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Gatti, D.; Viapiana, O.; Lippolis, I.; Braga, V.; Prizzi, R.; Rossini, M.; Adami, S. Intravenous bisphosphonate therapy increases radial width in adults with osteogenesis imperfecta. J. Bone Miner. Res. 2005, 20, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Lin, J. Bisphosphonates: A review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [Google Scholar] [CrossRef]
- Narayanan, P. Denosumab: A comprehensive review. South Asian J. Cancer 2013, 2, 272–277. [Google Scholar] [CrossRef]
- Yanling, J.; Jin, Y.; Levine, M.A.; Hoyer-Kuhn, H.; Ward, L.; Adachi, J.D. Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr. 2018, 107, 534–537. [Google Scholar] [CrossRef]
- Hoyer-Kuhn, H.; Franklin, J.; Allo, G.; Kron, M.; Netzer, C.; Eysel, P.; Hero, B.; Schoenau, E.; Semler, O. Safety and efficacy of denosumab in children with osteogenesis imperfect—A first prospective trial. J. Musculoskelet. Neuronal Interact. 2016, 16, 24–32. [Google Scholar]
- Bone, H.G.; Wagman, R.B.; Brandi, M.L.; Brown, J.P.; Chapurlat, R.; Cummings, S.R.; Czerwiński, E.; Fahrleitner-Pammer, A.; Kendler, D.L.; Lippuner, K.; et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: Results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. 2017, 5, 513–523. [Google Scholar] [CrossRef]
- Morello, R. Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018, 71, 294–312. [Google Scholar] [CrossRef]
- Marini, J.C.; Hopkins, E.; Glorieux, F.H.; Chrousos, G.P.; Reynolds, J.C.; Gundberg, C.M.; Reing, C.M. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfecta: High predictive value of the carboxyterminal propeptide of type I procollagen. J. Bone Miner. Res. 2003, 18, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Camacho, P.M.; Petak, S.M.; Binkley, N.; Clarke, B.L.; Harris, S.T.; Hurley, D.L.; Kleerekoper, M.; Lewiecki, E.M.; Miller, P.D.; Narula, H.S.; et al. Treatment algorithm postmenopausal osteoporosis. Endocr. Pract. 2016, 22, 1111–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orwoll, E.S.; Shapiro, J.; Veith, S.; Wang, Y.; Lapidus, J.; Vanek, C.; Reeder, J.L.; Keaveny, T.M.; Lee, D.C.; Mullins, M.A.; et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J. Clin. Investig. 2014, 124, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leali, P.T.; Balsano, M.; Maestretti, G.; Brusoni, M.; Amorese, V.; Ciurlia, E.; Andreozzi, M.; Caggiari, G.; Doria, C. Efficacy of teriparatide vs neridronate in adults with osteogenesis imperfecta type I: A prospective randomized international clinical study. Clin. Cases Miner. Bone Metab. 2017, 14, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Vahle, J.L.; Long, G.G.; Sandusky, G.; Westmore, M.; Ma, Y.L.; Sato, M. Bone neoplasms in F344 rats given teriparatide (rhPTH(1-34)) are dependent on duration of treatment and dose. Toxicol. Pathol. 2004, 32, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, F.H.; Devogelaer, J.P.; Durigova, M.; Goemaere, S.; Hemsley, S.; Jakob, F.; Junker, U.; Ruckle, J.; Seefried, L.; Winkle, P.J. BPS804 anti-sclerostin antibody in adults with moderate osteogenesis imperfecta: Results of a randomized phase 2a trial. J. Bone Miner. Res. 2017, 32, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Ebina, K.; Hirao, M.; Tsuboi, H.; Nagayama, Y.; Kashii, M.; Kaneshiro, S.; Miyama, A.; Nakaya, H.; Kunugiza, Y.; Okamura, G.; et al. Effects of prior osteoporosis treatment on early treatment response of romosozumab in pa-tients with postmenopausal osteoporosis. Bone 2020, 140, 115574. [Google Scholar] [CrossRef]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Cai, X.; Yang, W.; Gao, L.; Chen, L.; Wu, J.; Ji, L. Denosumab or romosozumab therapy and risk of cardiovascular events in patients with primary osteoporosis: Systematic review and meta-analysis. Bone 2020, 130, 115121. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Mundy, G.R. Role of transforming growth factor-beta in bone remodeling. Clin. Orthop. Relat. Res. 1990, 250, 261–276. [Google Scholar] [CrossRef]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Management of endocrine disease: Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef]
- Grafe, I.; Yang, T.; Alexander, S.; Homan, E.P.; Lietman, C.; Jiang, M.M.; Bertin, T.K.; Munivez, E.; Chen, Y.; Dawson, B.; et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014, 20, 670–675. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Daghlas, S.; Xie, Y.; Hulbert, A.M.; Pfeiffer, F.M.; Dallas, M.R.; Omosule, C.L.; Pearsall, R.S.; Dallas, S.L.; Phillips, C.L. Skeletal response to soluble activin receptor type IIB in mouse models of osteogenesis imperfecta. J. Bone Miner. Res. 2018, 33, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced ma-lignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- A Study in Adult Patients with Type I, III or IV Osteogenesis Imperfecta Treated with BPS804 (ASTEROID). Available online: https://clinicaltrials.gov/ct2/show/NCT03118570 (accessed on 30 August 2021).
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and im-proves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Riminucci, M.; Remoli, C.; Robey, P.G.; Bianco, P. Stem cells and bone diseases: New tools, new perspective. Bone 2015, 70, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.N.; Moschidou, D.; Abdulrazzak, H.; Kalirai, B.S.; Vanleene, M.; Osatis, S.; Shefelbine, S.J.; Horwood, N.J.; Marenzana, M.; De Coppi, P.; et al. Potential of human fetal chorionic stem cells for the treatment of osteogenesis imperfecta. Stem Cells Dev. 2014, 23, 262–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, E.M.; Prockop, D.J.; Fitzpatrick, L.A.; Koo, W.W.K.; Gordon, P.L.; Neel, M.; Sussman, M.; Orchard, P.; Marx, J.C.; Pyeritz, R.E.; et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat. Med. 1999, 5, 309–313. [Google Scholar] [CrossRef]
- Götherström, C.; Westgren, M.; Shaw, S.S.; Åström, E.; Biswas, A.; Byers, P.H.; Mattar, C.N.; Graham, G.E.; Taslimi, J.; Ewald, U.; et al. Pre- and postnatal Transplantation of Fetal mesenchymal stem cells in osteogenesis imperfecta: A two-center experience. Stem Cells Transl. Med. 2014, 3, 255–264. [Google Scholar] [CrossRef]
- Majka, M.; Janeczko, M.; Go′Zdzik, J.; Jarocha, D.; Auguściak-Duma, A.; Witecka, J.; Lesiak, M.; Koryciak-Komarska, H.; Sieroń, A.L.; Pietrzyk, J.J. Cell therapy of a patient with type III osteogenesis imperfecta caused by mutation in COL1A2 gene and unstable collagen type I. Open J. Genet. 2013, 3, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Deyle, D.R.; Khan, I.F.; Ren, G.; Wang, P.-R.; Kho, J.; Schwarze, U.; Russell, D.W. Normal collagen and bone production by gene-targeted human osteogenesis imperfecta iPSCs. Mol. Ther. 2012, 20, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, J.; Gioia, R.; Layrolle, P.; Lieubeau, B.; Heymann, D.; Rossi, A.; Marini, J.C.; Trichet, V.; Forlino, A. Allele-specific Col1a1 silencing reduces mutant collagen in fibroblasts from Brtl mouse, a model for classical osteogenesis imperfecta. Eur. J. Hum. Genet. 2013, 22, 667–674. [Google Scholar] [CrossRef]
- Calos, M. Genome editing techniques and their therapeutic applications. Clin. Pharmacol. Ther. 2016, 101, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-Y.; Ko, J.M.; Park, M.-H.; Koo, S.K. Generation of a patient-specific induced pluripotent stem cell line, KSCBi006-A, for osteogenesis imperfecta type I with the COL1A1, c.3162delT mutation. Stem Cell Res. 2019, 41, 101622. [Google Scholar] [CrossRef]
- Peng, G.-Y.; Lin, Y.; Li, J.-J.; Wang, Y.; Huang, H.-Y.; Shen, Z.-Y. The application of induced pluripotent stem cells in pathogenesis study and gene therapy for vascular disorders: Current progress and future challenges. Stem Cells Int. 2019, 2019, 9613258. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A comparative view on human somatic cell sources for iPSC generation. Stem Cells Int. 2014, 2014, 768391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Narayanan, K.; Lu, H.; Choo, Y.; Du, C.; Wiradharma, N.; Yang, Y.-Y.; Wan, A.C.A. Delivery of reprogramming factors into fibroblasts for generation of non-genetic induced pluripotent stem cells using a cationicbolaamphiphile as a non-viral vector. Biomaterials 2013, 34, 5336–5343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zi-Ying, Y.; Bian, G.-L.; Huang, H.-Y.; Shen, H.; Yang, J.-J.; Yang, Z.-Y.; Shen, Z.-Y. The combination of stem cells and tissue engineering: An advanced strategy for blood vessels regeneration and vascular disease treatment. Stem Cell Res. Ther. 2017, 8, 194. [Google Scholar] [CrossRef]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced pluripotent stem cells: Reprogramming platforms and applications in cell Replacement therapy. BioRes. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef]
- Fus-Kujawa, A.; Mendrek, B.; Trybus, A.; Bajdak-Rusinek, K.; Stepien, K.; Sieron, A. Potential of induced pluripotent stem cells for use in gene therapy: History, molecular bases, and medical perspectives. Biomoleculs 2021, 11, 699. [Google Scholar] [CrossRef]
- Dey, C.; Narayan, G.; Kumar, H.K.; Borgohain, M.; Lenka, N. Cell-penetrating peptides as a tool to deliver biologically active recombinant proteins to generate transgene-free induced pluripotent stem cells. Stud. Stem Cells Res. Ther. 2017, 3, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Fus-Kujawa, A.; Teper, P.; Botor, M.; Klarzyńska, K.; Sieroń, Ł.; Verbelen, B.; Smet, M.; Sieron, A.L.; Mendrek, B.; Kowalczuk, A. Functional star polymers as reagents for efficient nucleic acids delivery into HT-1080 cells. Int. J. Polym. Mater. 2021, 70, 356–370. [Google Scholar] [CrossRef]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.-H.; Pan, B.; Hu, Y.-J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef]
- Rauch, F.; Geng, Y.; Lamplugh, L.; Hekmatnejad, B.; Gaumond, M.-H.; Penney, J.; Yamanaka, Y.; Moffatt, P. Crispr-Cas9 engineered osteogenesis imperfecta type V leads to severe skeletal deformities and perinatal lethality in mice. Bone 2018, 107, 131–142. [Google Scholar] [CrossRef]
- Far, H.H.; Patria, Y.N.; Motazedian, A.; Elefanty, A.G.; Stanley, E.G.; Lamande, S.R.; Bateman, J.F. Generation of a heterozygous COL1A1 (c.3969_3970insT) osteogenesis im-perfecta mutation human iPSC line, MCRIi001-A-1, using CRISPR/Cas9 editing. Stem Cell Res. 2019, 37, 101449. [Google Scholar]
- Howden, S.; Far, H.H.; Motazedian, A.; Elefanty, A.; Stanley, E.G.; Lamande, S.; Bateman, J.F. The use of simultaneous reprogramming and gene correction to generate an osteogenesis imperfecta patient COL1A1 c. 3936 G > T iPSC line and an isogenic control iPSC line. Stem Cell Res. 2019, 38, 101453. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Rim, Y.; Park, N.; Nam, Y.; Ju, J. Restoration of osteogenesis by CRISPR/Cas9 genome editing of the mutated COL1A1 Gene in osteogenesis imperfecta. J. Clin. Med. 2021, 10, 3141. [Google Scholar] [CrossRef] [PubMed]
- Besio, R.; Iula, G.; Garibaldi, N.; Cipolla, L.; Sabbioneda, S.; Biggiogera, M.; Marini, J.C.; Rossi, A.; Forlino, A. 4-PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Besio, R.; Garibaldi, N.; Leoni, L.; Cipolla, L.; Sabbioneda, S.; Biggiogera, M.; Mottes, M.; Aglan, M.; Otaify, G.A.; Temtamy, S.A.; et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. Dis. Models Mech. 2019, 12, dmm038521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gioia, R.; Tonelli, F.; Ceppi, I.; Biggiogera, M.; Leikin, S.; Fisher, S.; Tenedini, E.; Yorgan, T.A.; Schinke, T.; Tian, K.; et al. The chaperone activity of 4PBA ameliorates the skeletal phenotype of Chihuahua, a zebrafish model for dominant osteogenesis imperfecta. Hum. Mol. Genet. 2017, 26, 2897–2911. [Google Scholar] [CrossRef] [Green Version]
- Garibaldi, N.; Contento, B.M.; Babini, G.; Morini, J.; Siciliani, S.; Biggiogera, M.; Raspanti, M.; Marini, J.C.; Rossi, A.; Forlino, A.; et al. Targeting cellular stress in vitro improves osteoblast homeostasis, matrix collagen content and mineralization in two murine models of osteogenesis imperfecta. Matrix Biol. 2021, 98, 1–20. [Google Scholar] [CrossRef]
- Duangchan, T.; Tawonsawatruk, T.; Angsanuntsukh, C.; Trachoo, O.; Hongeng, S.; Kitiyanant, N.; Supokawej, A. Amelioration of osteogenesis in iPSC-derived mesenchymal stem cells from osteogenesis imperfecta patients by endoplasmic reticulum stress inhibitor. Life Sci. 2021, 278, 119628. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Type of OI | Inheritance Mode | Mutated Gene(s) | O(MIM) Number (Gene) | Clinical Characteristics |
|---|---|---|---|---|
| Osteogenesis imperfecta type I | AD | COL1A1 | 120150 | Mild, non-deforming, with normal stature, increased bone fragility, blue-grey sclerae, hearing loss [5] |
| COL1A2 | 120160 | |||
| Osteogenesis imperfecta type II | AD | COL1A1 | 120150 | Lethal in the perinatal period [5] |
| COL1A2 | 120160 | |||
| AR | CRTAP | 605497 | Severe to lethal [6,7,8] | |
| P3H1 | 610339 | |||
| Osteogenesis imperfecta type III | AD | COL1A1 | 120150 | Progressively deforming [5] |
| COL1A2 | 120160 | |||
| IFITM5 | 614757 | Mild to moderate [9] | ||
| AR | SERPINF1 | 172860 | Moderate to severe, scoliosis [10] | |
| CRTAP | 605497 | Severe to lethal [6,7,8] | ||
| P3H1 | 610339 | |||
| SERPINH1 | 600943 | Severe to lethal [11] | ||
| AR | FKBP10 | 607063 | Broad-spectrum, includes OI, Bruck syndrome, Kuskokwim syndrome (joint contractures at birth, short stature) [12] | |
| AR | BMP1 | 112264 | Severe; high bone mass [13] | |
| WNT1 | 164820 | Moderately severe [14] | ||
| Osteogenesis imperfecta type IV | AD | COL1A1 | 120150 | Moderately deforming [5] |
| COL1A2 | 120160 | |||
| WNT1 | 164820 | Moderately severe [14] | ||
| IFITM5 | 614757 | Mild to moderate [9] | ||
| AR | FKBP10 | 607063 | Progressive, deforming [12] | |
| SP7 | 606633 | Moderate [15] | ||
| Osteogenesis imperfecta type V | AD | IFITM5 | 614757 | Mild to moderate (moderately deforming) with calcification of interosseous membranes and/or hypertrophic callus [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botor, M.; Fus-Kujawa, A.; Uroczynska, M.; Stepien, K.L.; Galicka, A.; Gawron, K.; Sieron, A.L. Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules 2021, 11, 1493. https://doi.org/10.3390/biom11101493
Botor M, Fus-Kujawa A, Uroczynska M, Stepien KL, Galicka A, Gawron K, Sieron AL. Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules. 2021; 11(10):1493. https://doi.org/10.3390/biom11101493
Chicago/Turabian StyleBotor, Malwina, Agnieszka Fus-Kujawa, Marta Uroczynska, Karolina L. Stepien, Anna Galicka, Katarzyna Gawron, and Aleksander L. Sieron. 2021. "Osteogenesis Imperfecta: Current and Prospective Therapies" Biomolecules 11, no. 10: 1493. https://doi.org/10.3390/biom11101493
APA StyleBotor, M., Fus-Kujawa, A., Uroczynska, M., Stepien, K. L., Galicka, A., Gawron, K., & Sieron, A. L. (2021). Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules, 11(10), 1493. https://doi.org/10.3390/biom11101493