
Advances in Proteasome Enhancement by Small Molecules



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Human Proteasome
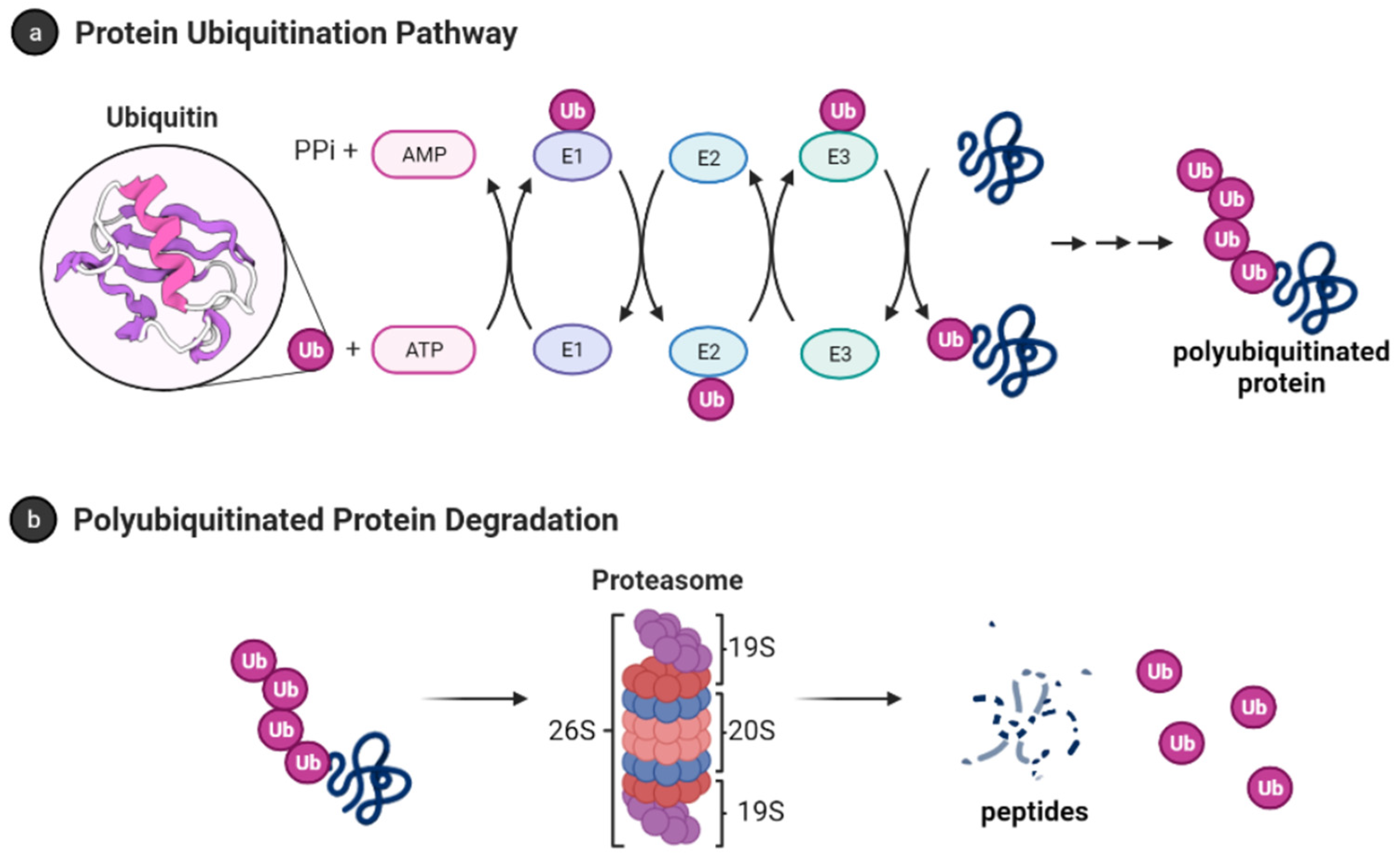
1.2. Ubiquitin-Proteasome System
1.2.1. Ubiquitin
1.2.2. The 26S Proteasome
1.3. The 20S Proteasome or Core Particle
1.4. Small Molecule Regulation of Proteasome Function
2. Proteasome Activity and Diseases
2.1. Aging
2.2. Neurodegenerative Diseases
2.2.1. Parkinson’s Disease (PD)
2.2.2. Alzheimer’s Disease (AD)
2.2.3. Huntington’s Disease (HD)
2.2.4. Amyotrophic Lateral Sclerosis (ALS)
3. Small Molecule Enhancers of 26S Proteasome Activity
3.1. Indirect Activation of 26S Proteasome
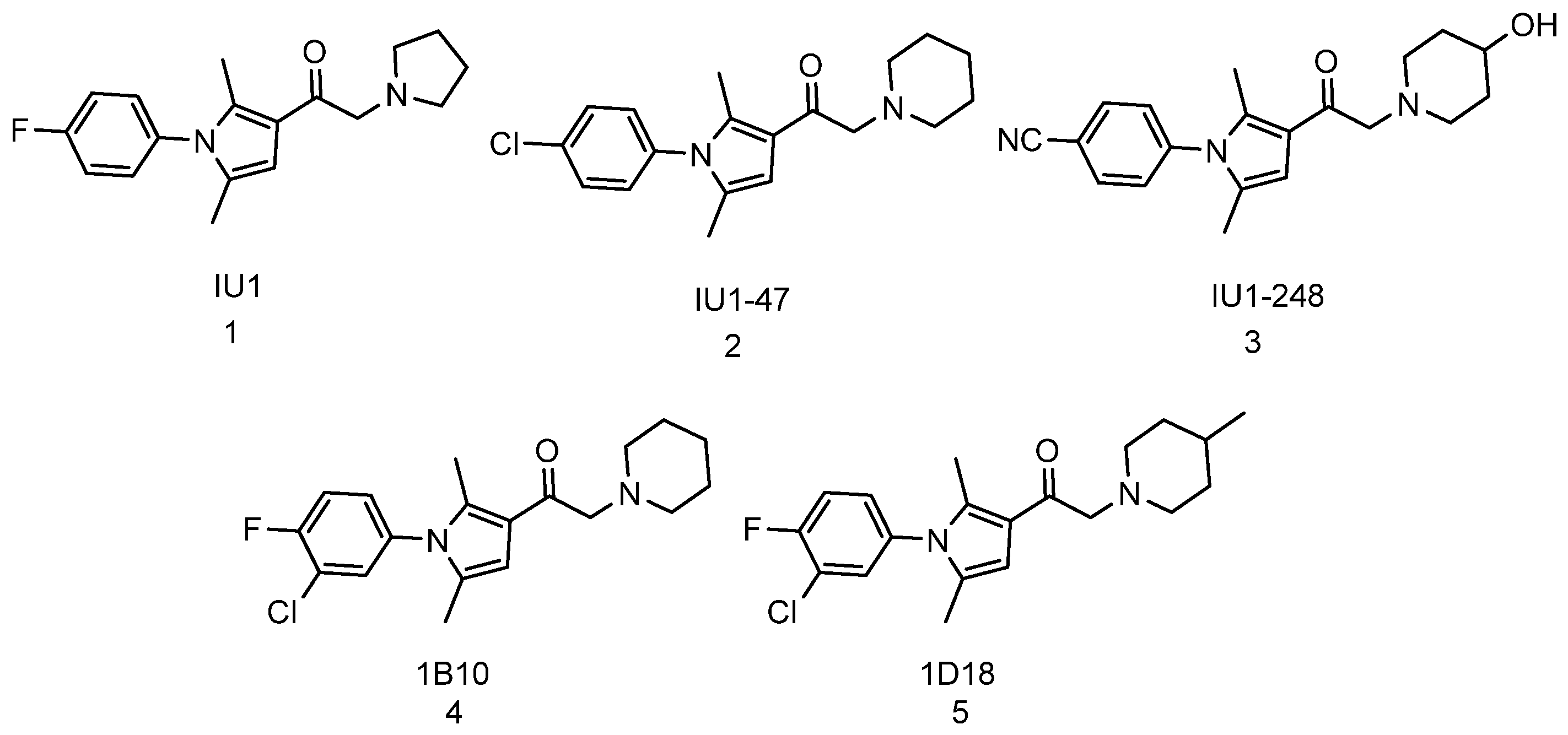
3.1.1. Inhibition of Deubiquitinase
3.1.2. Modulation of cAMP-Dependent Protein Kinase A (PKA) and cGMP-Dependent Protein Kinase G
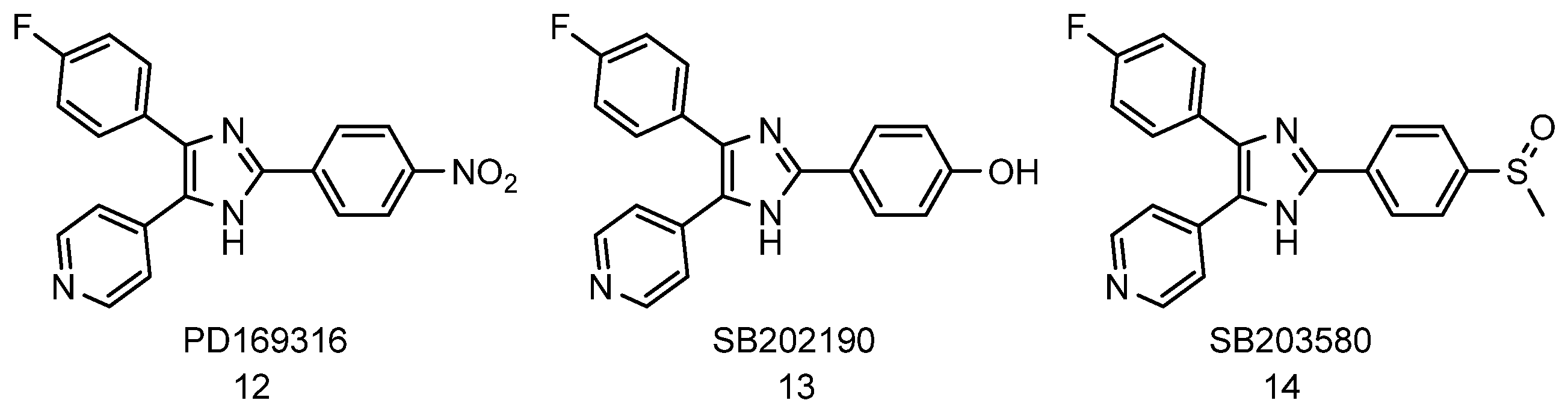
3.1.3. Inhibition of p38 Mitogen-Activated Protein Kinase (MAPK)
3.1.4. Proteasome Activation by Genetic Manipulation
4. Small Molecule Enhancers of 20S Proteasome Activity
4.1. Sodium Dodecyl Sulfate (SDS)
4.2. Natural Product-Based Activators
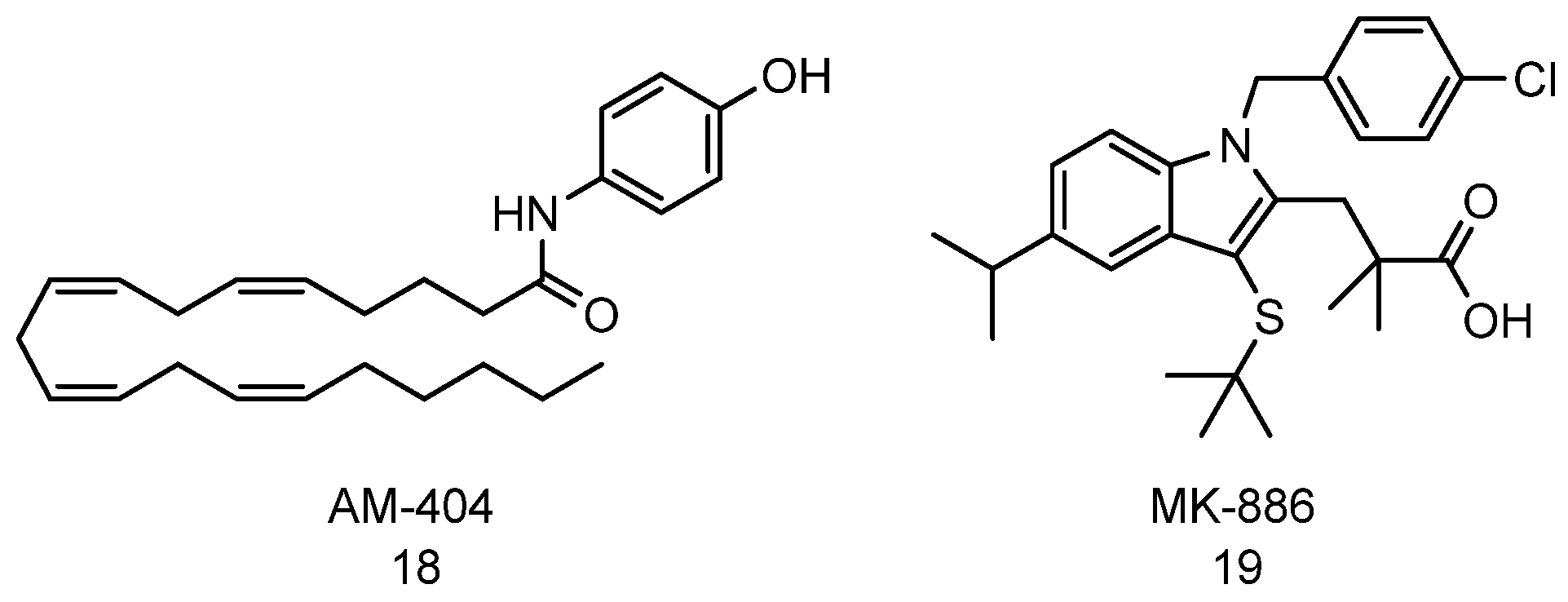
4.3. AM-404 and MK-886
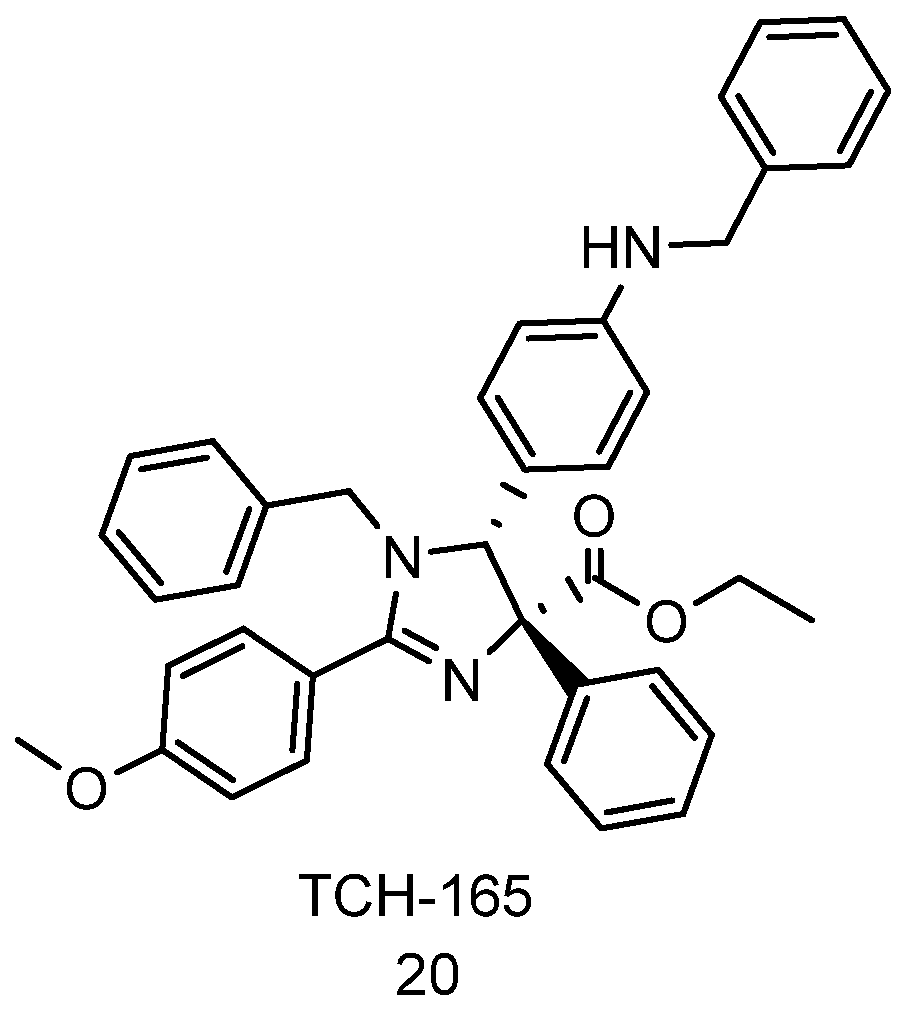
4.4. Imidazolines
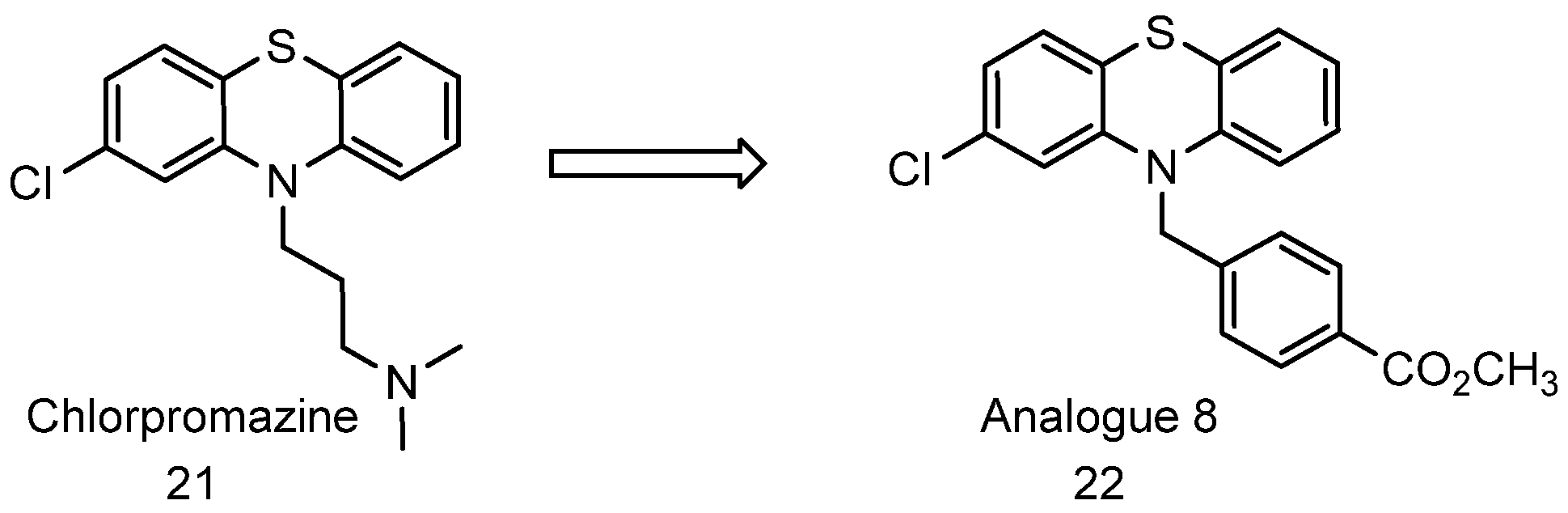
4.5. Chlorpromazines
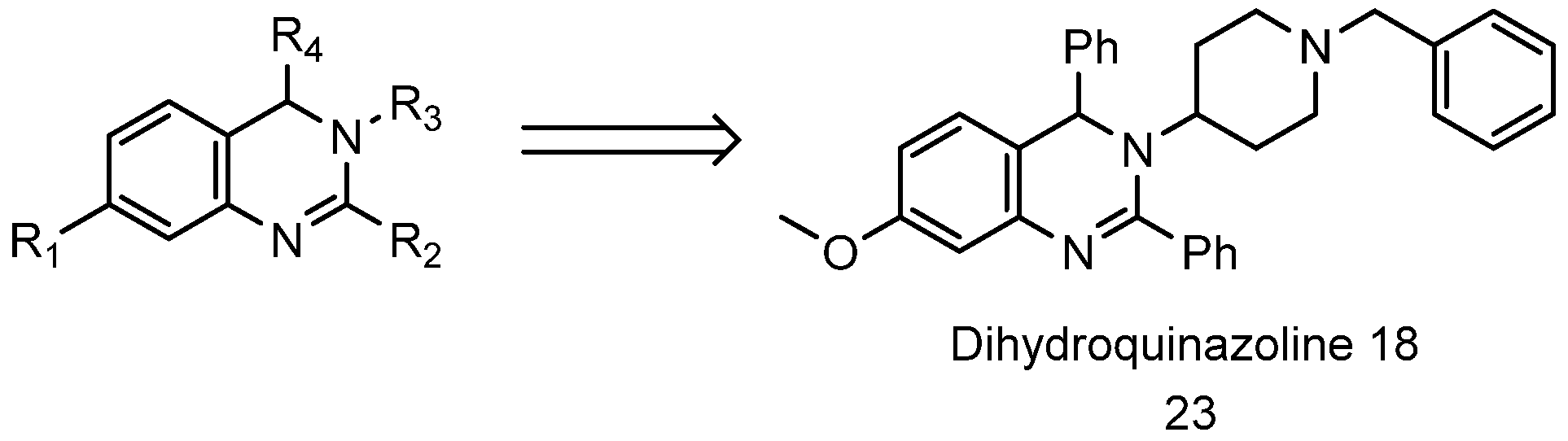
4.6. Dihydroquinazolines
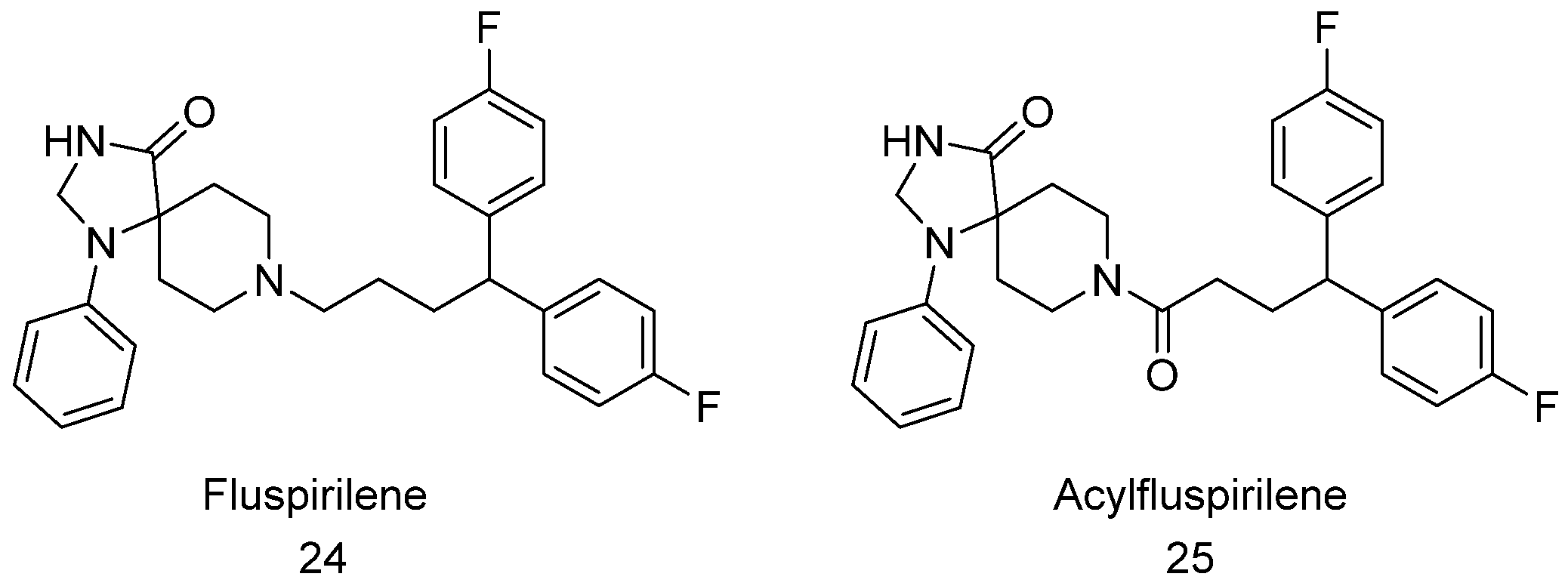
4.7. Fluspirilene and Acylfluspirilene
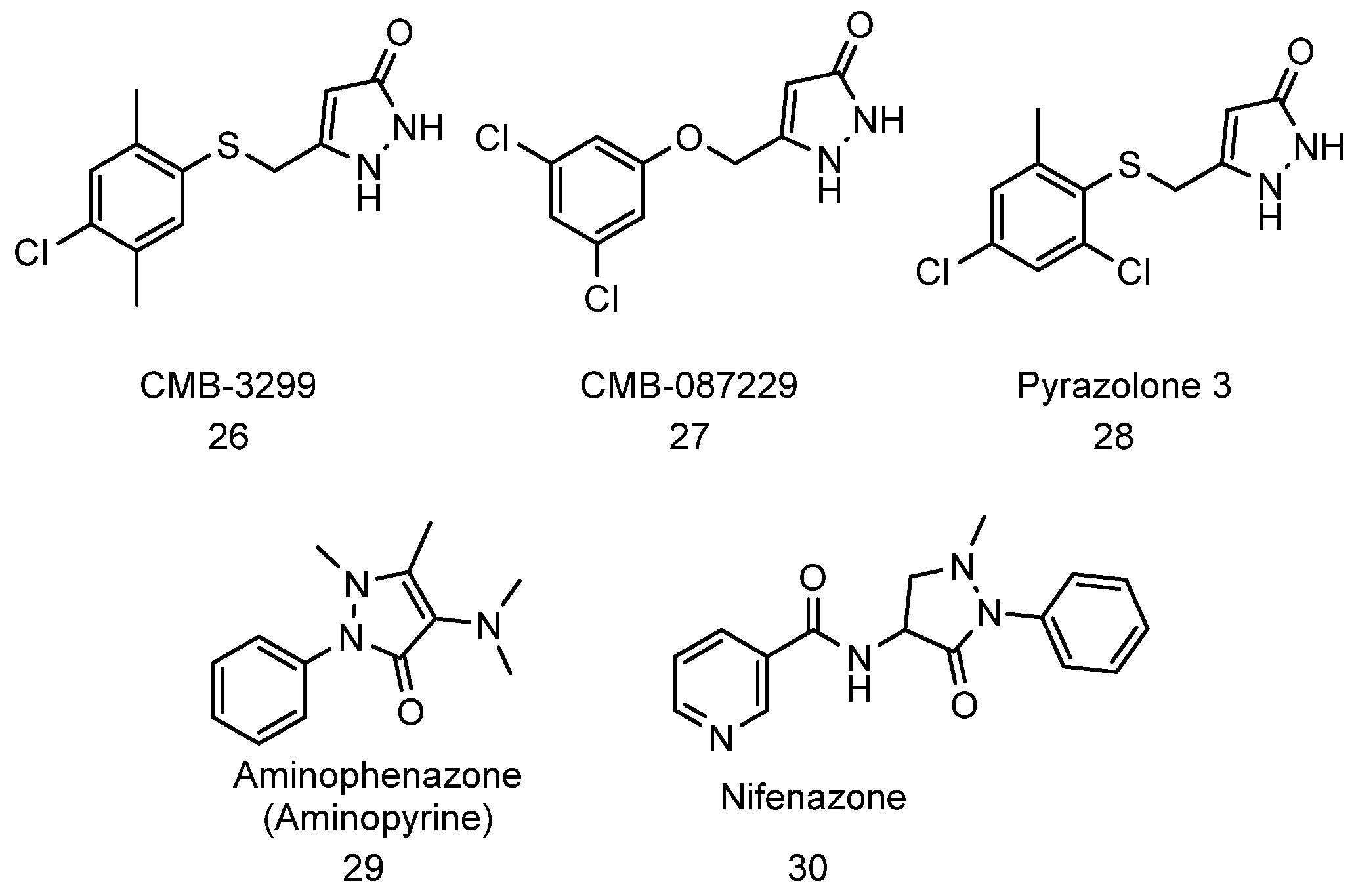
4.8. Pyrazolones
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hetz, C.; Glimcher, L.H. Protein homeostasis networks in physiology and disease. Curr. Opin. Cell. Biol. 2011, 23, 123–125. [Google Scholar] [CrossRef] [Green Version]
- McNaught, K.S.P.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin–proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genomics 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPlante, G.; Zhang, W. Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers 2021, 13, 3079. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, S.; Memariani, Z.; El-Senduny, F.F.; Sanadgol, N.; Golab, F.; Katebi, M.; Abdolghaffari, A.H.; Farzaei, M.H.; Abdollahi, M. Targeting Ubiquitin-Proteasome Pathway by Natural Products: Novel Therapeutic Strategy for Treatment of Neurodegenerative Diseases. Front. Physiol. 2020, 11, 361. [Google Scholar] [CrossRef]
- Rao, G.; Croft, B.; Teng, C.; Awasthi, V. Ubiquitin-Proteasome System in Neurodegenerative Disorders. J. Drug Metab. Toxicol. 2015, 6, 187. [Google Scholar] [PubMed] [Green Version]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis 2010, 15, 1292–1311. [Google Scholar] [CrossRef] [Green Version]
- Njomen, E.; Tepe, J.J. Proteasome activation as a new therapeutic approach to target proteotoxic disorders. J. Med. Chem. 2019, 62, 6469–6481. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Akopian, T.N.; Woo, K.M.; Goldberg, A.L. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 1999, 274, 3363–3371. [Google Scholar] [CrossRef] [Green Version]
- Kumar Deshmukh, F.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The Contribution of the 20S Proteasome to Proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adapted from “Ubiquitin Proteasome System”, by BioRender.com. Available online: https://app.biorender.com/biorender-templates (accessed on 19 October 2021).
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell. Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- hrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Dimova, N.V.; Hathaway, N.A.; Lee, B.-H.; Kirkpatrick, D.S.; Berkowitz, M.L.; Gygi, S.P.; Finley, D.; King, R.W. APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat. Cell Biol. 2012, 14, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Shabek, N.; Herman-Bachinsky, Y.; Buchsbaum, S.; Lewinson, O.; Haj-Yahya, M.; Hejjaoui, M.; Lashuel, H.A.; Sommer, T.; Brik, A.; Ciechanover, A. The Size of the Proteasomal Substrate Determines Whether Its Degradation Will Be Mediated by Mono- or Polyubiquitylation. Mol. Cell 2012, 48, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Cejka, Z.; Harris, J.R.; Kleinschmidt, J.A.; Baumeister, W. Structural features of the 26S proteasome complex. J. Mol. Biol. 1993, 234, 932–937. [Google Scholar] [CrossRef]
- da Fonseca, P.C.; Morris, E.P. Structure of the human 26S proteasome: Subunit radial displacements open the gate into the proteolytic core. J. Biol. Chem. 2008, 283, 23305–23314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlowski, M.; Wilk, S. Catalytic Activities of the 20S Proteasome, a Multicatalytic Proteinase Complex. Arch. Biochem. Biophys. 2000, 383, 1–16. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci 2016, 41, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Andres, H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis–driven translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef] [Green Version]
- Eisele, M.R.; Reed, R.G.; Rudack, T.; Schweitzer, A.; Beck, F.; Nagy, I.; Pfeifer, G.; Plitzko, J.M.; Baumeister, W.; Tomko, R.J.; et al. Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Rep. 2018, 24, 1301–1315.e1305. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Fu, Z.; Xu, C.; Wang, Y.; Wang, Y.; Li, J.; Kong, L.; Chen, J.; Li, N.; Zhang, R.; et al. High-resolution cryo-EM structure of the proteasome in complex with ADP-AlFx. Cell Res. 2017, 27, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Luan, B.; Wu, J.; Shi, Y. An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 2016, 23, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, J.; Lu, Y.; Ma, Y.-B.; Lee, B.-H.; Yu, Z.; Ouyang, Q.; Finley, D.J.; Kirschner, M.W.; Mao, Y. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, 12991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Xu, C.; Sahu, I.; Wang, Y.; Fu, Z.; Huang, M.; Wong, C.C.L.; Glickman, M.H.; Cong, Y. Structural Snapshots of 26S Proteasome Reveal Tetraubiquitin-Induced Conformations. Mol. Cell 2019, 73, 1150–1161.e1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M.; Ditzel, L.; Groll, M.; Hartmann, C.; Huber, R. The proteasome. Annu Rev Biophys Biomol Struct 1999, 28, 295–317. [Google Scholar] [CrossRef]
- Huber, E.M.; Heinemeyer, W.; Li, X.; Arendt, C.S.; Hochstrasser, M.; Groll, M. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun. 2016, 7, 10900. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chen, C.H. Proteasome regulators: Activators and inhibitors. Curr. Med. Chem. 2009, 16, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Bruna, J.; Udina, E.; Alé, A.; Vilches, J.J.; Vynckier, A.; Monbaliu, J.; Silverman, L.; Navarro, X. Neurophysiological, histological and immunohistochemical characterization of bortezomib-induced neuropathy in mice. Exp. Neurol. 2010, 223, 599–608. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Russo, S.M.; Tepper, J.E.; Baldwin, A.S., Jr.; Liu, R.; Adams, J.; Elliott, P.; Cusack, J.C., Jr. Enhancement of radiosensitivity by proteasome inhibition: Implications for a role of NF-kappaB. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 183–193. [Google Scholar] [CrossRef]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Crowl Bancroft, C.; Sausville, E.; Adams, J.; Elliott, P.; Van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 1419–1428. [Google Scholar]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.; Waldmann, T.A. Proteasome inhibitor PS-341, a potential therapeutic agent for adult T-cell leukemia. Cancer Res. 2002, 62, 1083–1086. [Google Scholar]
- Ma, M.H.; Yang, H.H.; Parker, K.; Manyak, S.; Friedman, J.M.; Altamirano, C.; Wu, Z.Q.; Borad, M.J.; Frantzen, M.; Roussos, E.; et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin. Cancer Res. 2003, 9, 1136–1144. [Google Scholar]
- Shah, S.A.; Potter, M.W.; McDade, T.P.; Ricciardi, R.; Perugini, R.A.; Elliott, P.J.; Adams, J.; Callery, M.P. 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J. Cell. Biochem. 2001, 82, 110–122. [Google Scholar] [CrossRef]
- Yang, Y.; Ikezoe, T.; Saito, T.; Kobayashi, M.; Koeffler, H.P.; Taguchi, H. Proteasome inhibitor PS-341 induces growth arrest and apoptosis of non-small cell lung cancer cells via the JNK/c-Jun/AP-1 signaling. Cancer Sci. 2004, 95, 176–180. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.A.; McConkey, D.J. The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res. 2003, 63, 7338–7344. [Google Scholar]
- Li, B.; Dou, Q.P. Bax degradation by the ubiquitin/proteasome-dependent pathway: Involvement in tumor survival and progression. Proc. Natl. Acad. Sci. USA 2000, 97, 3850–3855. [Google Scholar] [CrossRef] [Green Version]
- Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J. Biol. Chem. 2000, 275, 21648–21652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Goldberg, A.L. Multiple myeloma cells are exceptionally sensitive to heat shock, which overwhelms their proteostasis network and induces apoptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 21588. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; Münch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Aliabadi, F.; Sohrabi, B.; Mostafavi, E.; Pazoki-Toroudi, H.; Webster, T.J. Ubiquitin–proteasome system and the role of its inhibitors in cancer therapy. Open Biol. 2021, 11, 200390. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, G.S.; Torcun, C.C.; Grune, T.; Ozer, N.K.; Karademir, B. Proteasome inhibitors in cancer therapy: Treatment regimen and peripheral neuropathy as a side effect. Free Radic. Biol. Med. 2017, 103, 1–13. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Narayanan, S.; Cai, C.-Y.; Assaraf, Y.G.; Guo, H.-Q.; Cui, Q.; Wei, L.; Huang, J.-J.; Ashby, C.R.; Chen, Z.-S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updates 2020, 48, 100663. [Google Scholar] [CrossRef] [PubMed]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Sherman, D.J.; Li, J. Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbell, G.E.; Tepe, J.J. Natural product scaffolds as inspiration for the design and synthesis of 20S human proteasome inhibitors. RSC Chem. Biol. 2020, 1, 305–332. [Google Scholar] [CrossRef]
- Jones, C.L.; Tepe, J.J. Proteasome Activation to Combat Proteotoxicity. Molecules 2019, 24, 2841. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Njomen, E.; Sjogren, B.; Dexheimer, T.S.; Tepe, J.J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017, 12, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Njomen, E.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.; Tepe, J.J. Small Molecule Modulation of Proteasome Assembly. Biochemistry 2018, 57, 4214–4224. [Google Scholar] [CrossRef]
- Trippier, P.C.; Zhao, K.T.; Fox, S.G.; Schiefer, I.T.; Benmohamed, R.; Moran, J.; Kirsch, D.R.; Morimoto, R.I.; Silverman, R.B. Proteasome Activation is a Mechanism for Pyrazolone Small Molecules Displaying Therapeutic Potential in Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2014, 5, 823–829. [Google Scholar] [CrossRef]
- Trader, D.J.; Simanski, S.; Dickson, P.; Kodadek, T. Establishment of a suite of assays that support the discovery of proteasome stimulators. Biochim. Biophys. Acta 2017, 1861, 892–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene Expression Profile of Aging and Its Retardation by Caloric Restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.-L.; Lundberg, K.C.; Humphries, K.M.; Sadek, H.A.; Szweda, P.A.; Friguet, B.; Szweda, L.I. Oxidative Modification and Inactivation of the Proteasome during Coronary Occlusion/Reperfusion*. J. Biol. Chem. 2001, 276, 30057–30063. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S Proteasome Complex During Oxidative Stress. Sci. Signal. 2010, 3, ra88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonoki, A.; Kuranaga, E.; Tomioka, T.; Hamazaki, J.; Murata, S.; Tanaka, K.; Miura, M. Genetic Evidence Linking Age-Dependent Attenuation of the 26S Proteasome with the Aging Process. Mol. Cell. Biol. 2009, 29, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Bajorek, M.; Finley, D.; Glickman, M.H. Proteasome Disassembly and Downregulation Is Correlated with Viability during Stationary Phase. Curr. Biol. 2003, 13, 1140–1144. [Google Scholar] [CrossRef]
- Kayed, R.; Dettmer, U.; Lesné, S.E. Soluble endogenous oligomeric α-synuclein species in neurodegenerative diseases: Expression, spreading, and cross-talk. J. Parkinsons Dis. 2020, 10, 791–818. [Google Scholar] [CrossRef] [PubMed]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.; Honig, L.S. Role of amyloid-β and tau proteins in Alzheimer’s disease: Confuting the amyloid cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Ghag, G.; Bhatt, N.; Cantu, D.V.; Guerrero-Munoz, M.J.; Ellsworth, A.; Sengupta, U.; Kayed, R. Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci. 2018, 27, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
- Forloni, G.; Balducci, C. Alzheimer’s disease, oligomers, and inflammation. J. Alzheimer’s Dis. 2018, 62, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Sengupta, U.; Gerson, J.E.; Kayed, R. α-Synuclein oligomers induce a unique toxic tau strain. Biol. Psychiatry 2018, 84, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelsson, M. Alpha-synuclein oligomers—neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Caárdenas-Aguayo, M.a.d.C.; Goόmez-Virgilio, L.; DeRosa, S.; Meraz-Ríos, M.A. The role of tau oligomers in the onset of Alzheimer’s disease neuropathology. ACS Chem. Neurosci. 2014, 5, 1178–1191. [Google Scholar] [CrossRef]
- Katzmarski, N.; Ziegler-Waldkirch, S.; Scheffler, N.; Witt, C.; Abou-Ajram, C.; Nuscher, B.; Prinz, M.; Haass, C.; Meyer-Luehmann, M. Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathol. 2020, 30, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M. Could a common mechanism of protein degradation impairment underlie many neurodegenerative diseases? J. Exp. Neurosci. 2018, 12, 1179069518794675. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Murillo, M.C.; Sengupta, U.; Barrett, A.; Kayed, R. Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol. Neurodegener. 2018, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau oligomers as pathogenic seeds: Preparation and propagation in vitro and in vivo. In Tau Protein; Humana Press: New York, NY, USA, 2017; pp. 141–157. [Google Scholar]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Gerson, J.E.; Mudher, A.; Kayed, R. Potential mechanisms and implications for the formation of tau oligomeric strains. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 482–496. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Cecarini, V.; Bonfili, L.; Amici, M.; Angeletti, M.; Keller, J.N.; Eleuteri, A.M. Amyloid peptides in different assembly states and related effects on isolated and cellular proteasomes. Brain Res. 2008, 1209, 8–18. [Google Scholar] [CrossRef]
- Díaz-Hernández, M.; Valera, A.G.; Morán, M.A.; Gómez-Ramos, P.; Alvarez-Castelao, B.; Castaño, J.G.; Hernández, F.; Lucas, J.J. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 2006, 98, 1585–1596. [Google Scholar] [CrossRef]
- Gregori, L.; Fuchs, C.; Figueiredo-Pereira, M.E.; Van Nostrand, W.E.; Goldgaber, D. Amyloid β-Protein Inhibits Ubiquitin-dependent Protein Degradation in Vitro (∗). J. Biol. Chem. 1995, 270, 19702–19708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindersson, E.; Beedholm, R.; Højrup, P.; Moos, T.; Gai, W.; Hendil, K.B.; Jensen, P.H. Proteasomal inhibition by α-synuclein filaments and oligomers. J. Biol. Chem. 2004, 279, 12924–12934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Oh, S.; Hong, H.S.; Hwang, E.; Sim, H.J.; Lee, W.; Shin, S.J.; Mook-Jung, I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 2005, 126, 1292–1299. [Google Scholar] [CrossRef]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef]
- Deriziotis, P.; André, R.; Smith, D.M.; Goold, R.; Kinghorn, K.J.; Kristiansen, M.; Nathan, J.A.; Rosenzweig, R.; Krutauz, D.; Glickman, M.H. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J. 2011, 30, 3065–3077. [Google Scholar] [CrossRef] [PubMed]
- Deriziotis, P.; Tabrizi, S.J. Prions and the proteasome. Biochim. Biophys. Acta 2008, 1782, 713–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.; Menéndez-Benito, V.; Dantuma, N.P. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by α-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Papanikolopoulou, K.; Skoulakis, E. Altered proteostasis in neurodegenerative tauopathies. In Proteostasis and Disease; Barrio, R., Sutherland, J.D., Rodriguez, M.S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 177–194. [Google Scholar]
- Choi, W.H.; De Poot, S.A.; Lee, J.H.; Kim, J.H.; Han, D.H.; Kim, Y.K.; Finley, D.; Lee, M.J. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J. Proteasome activation by small molecules. Cell Chem. Biol. 2017, 24, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Leestemaker, Y.; Ovaa, H. Tools to investigate the ubiquitin proteasome system. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Fiolek, T.J.; Magyar, C.L.; Wall, T.J.; Davies, S.B.; Campbell, M.V.; Savich, C.J.; Tepe, J.J.; Mosey, R.A. Dihydroquinazolines enhance 20S proteasome activity and induce degradation of alpha-synuclein, an intrinsically disordered protein associated with neurodegeneration. Bioorg. Med. Chem. Lett. 2021, 36, 127821. [Google Scholar] [CrossRef]
- Coleman, R.A.; Muli, C.S.; Zhao, Y.; Bhardwaj, A.; Newhouse, T.R.; Trader, D.J. Analysis of chain length, substitution patterns, and unsaturation of AM-404 derivatives as 20S proteasome stimulators. Bioorg. Med. Chem. Lett. 2019, 29, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Trader, D.J. Development and Application of a Sensitive Peptide Reporter to Discover 20S Proteasome Stimulators. ACS Comb. Sci. 2018, 20, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Giżynńska, M.; Witkowska, J.; Karpowicz, P.; Rostankowski, R.; Chocron, E.S.; Pickering, A.M.; Osmulski, P.; Gaczynska, M.; Jankowska, E.b. Proline-and arginine-rich peptides as flexible allosteric modulators of human proteasome activity. J. Med. Chem. 2019, 62, 359–370. [Google Scholar] [CrossRef]
- Njomen, E.; Lansdell, T.; Vanecek, A.; Benham, V.; Bernard, M.; Yang, Y.-T.; Schall, P.; Isaac, D.; Alkharabsheh, O.; Al-Janadi, A. Enhancing c-MYC degradation via 20S proteasome activation induces in vivo anti-tumor efficacy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. 2015, 29, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Gonos, E. Proteasome activation as a novel anti-aging strategy. Free Radic. Biol. Med. 2014, 75, S7. [Google Scholar] [CrossRef] [PubMed]
- Fiolek, T.J.; Keel, K.L.; Tepe, J.J. Fluspirilene Analogs Activate the 20S Proteasome and Overcome Proteasome Impairment by Intrinsically Disordered Protein Oligomers. ACS Chem. Neurosci. 2021, 12, 1438–1448. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njomen, E.; Tepe, J.J. Regulation of Autophagic Flux by the 20S Proteasome. Cell Chem. Biol. 2019, 26, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Reuven, N.; Shaul, Y. 20S proteasomes and protein degradation "by default". Bioessays 2006, 28, 844–849. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Höhn, T.J.; Grune, T. The proteasome and the degradation of oxidized proteins: Part III-Redox regulation of the proteasomal system. Redox Biol. 2014, 2, 388–394. [Google Scholar] [PubMed] [Green Version]
- Chondrogianni, N.; Petropoulos, I.; Grimm, S.; Georgila, K.; Catalgol, B.; Friguet, B.; Grune, T.; Gonos, E.S. Protein damage, repair and proteolysis. Mol. Aspects Med. 2014, 35, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Myers, N.; Olender, T.; Savidor, A.; Levin, Y.; Reuven, N.; Shaul, Y. The Disordered Landscape of the 20S Proteasome Substrates Reveals Tight Association with Phase Separated Granules. Proteomics 2018, 18, e1800076. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. The nanny model for IDPs. Nat. Chem. Biol. 2009, 5, 778–781. [Google Scholar] [CrossRef]
- Oliver, C.N.; Ahn, B.W.; Moerman, E.J.; Goldstein, S.; Stadtman, E.R. Age-related changes in oxidized proteins. J. Biol. Chem. 1987, 262, 5488–5491. [Google Scholar] [CrossRef]
- Carrard, G.; Dieu, M.; Raes, M.; Toussaint, O.; Friguet, B. Impact of ageing on proteasome structure and function in human lymphocytes. Int. J. Biochem. Cell Biol. 2003, 35, 728–739. [Google Scholar] [CrossRef]
- Keller, J.N.; Huang, F.F.; Markesbery, W.R. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 2000, 98, 149–156. [Google Scholar] [CrossRef]
- Giannini, C.; Kloß, A.; Gohlke, S.; Mishto, M.; Nicholson, T.P.; Sheppard, P.W.; Kloetzel, P.M.; Dahlmann, B. Poly-Ub-substrate-degradative activity of 26S proteasome is not impaired in the aging rat brain. PLoS ONE 2013, 8, e64042. [Google Scholar] [CrossRef] [Green Version]
- Merker, K.; Sitte, N.; Grune, T. Hydrogen peroxide-mediated protein oxidation in young and old human MRC-5 fibroblasts. Arch. Biochem. Biophys. 2000, 375, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Sitte, N.; Merker, K.; von Zglinicki, T.; Grune, T. Protein oxidation and degradation during proliferative senescence of human MRC-5 fibroblasts. Free Radic. Biol. Med. 2000, 28, 701–708. [Google Scholar] [CrossRef]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2014, 23, 239–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deger, J.M.; Gerson, J.E.; Kayed, R. The interrelationship of proteasome impairment and oligomeric intermediates in neurodegeneration. Aging Cell 2015, 14, 715–724. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Goold, R.; Andre, R.; Devoy, A.; Ortega, Z.; Moonga, J.; Linehan, J.M.; Brandner, S.; Lucas, J.J.; Collinge, J.; et al. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol. 2016, 131, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Otero, A.; Betancor, M.; Erana, H.; Fernandez Borges, N.; Lucas, J.J.; Badiola, J.J.; Castilla, J.; Bolea, R. Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease. Int. J. Mol. Sci. 2021, 22, 465. [Google Scholar] [CrossRef]
- Parkinson’s Disease Foundation. Available online: https://www.parkinson.org/Understanding-Parkinsons/Statistics (accessed on 17 October 2021).
- Moore, D.J.; Dawson, V.L.; Dawson, T.M. Role for the ubiquitin-proteasome system in Parkinson’s disease and other neurodegenerative brain amyloidoses. Neuromolecular Med. 2003, 4, 95–108. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Perl, D.P.; Brownell, A.L.; Olanow, C.W. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 2004, 56, 149–162. [Google Scholar] [CrossRef]
- Matsuda, N.; Tanaka, K. Does Impairment of the Ubiquitin-Proteasome System or the Autophagy-Lysosome Pathway Predispose Individuals to Neurodegenerative Disorders such as Parkinson’s Disease? J. Alzheimer’s Dis. 2010, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2012, 13, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FastStats-Leading Causes of Death. Available online: https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm (accessed on 17 October 2021).
- 2019 Alzheimer’s disease facts and figures. Alzheimer’s & Dement. 2019, 15, 321–387.
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Lourenco, M.; Oliveira, M.; De Felice, F. Soluble amyloid-b oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Lopez Salon, M.; Pasquini, L.; Besio Moreno, M.; Pasquini, J.M.; Soto, E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003, 180, 131–143. [Google Scholar] [CrossRef]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Saunders, A.J. The role of ubiquitin-proteasome in the metabolism of amyloid precursor protein (APP): Implications for novel therapeutic strategies for Alzheimer’s disease. Discov. Med. 2014, 18, 41–50. [Google Scholar] [PubMed]
- Riederer, B.M.; Leuba, G.; Vernay, A.; Riederer, I.M. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp. Biol. Med. 2011, 236, 268–276. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Snell, R.G.; MacMillan, J.C.; Cheadle, J.P.; Fenton, I.; Lazarou, L.P.; Davies, P.; MacDonald, M.E.; Gusella, J.F.; Harper, P.S.; Shaw, D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993, 4, 393–397. [Google Scholar] [CrossRef]
- Nance, M.A.; Myers, R.H. Juvenile onset Huntington’s disease-clinical and research perspectives. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 153–157. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Kalchman, M.A.; Graham, R.K.; Xia, G.; Koide, H.B.; Hodgson, J.G.; Graham, K.C.; Goldberg, Y.P.; Gietz, R.D.; Pickart, C.M.; Hayden, M.R. Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J. Biol. Chem. 1996, 271, 19385–19394. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.; Sonntag, K.C.; Kim, W.; Cattaneo, E.; Isacson, O. Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS One 2007, 2, e238. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O′Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Peter, E.A.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.-M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar Hans, A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172, 696–705.e612. [Google Scholar] [CrossRef] [Green Version]
- Kato, S. Amyotrophic lateral sclerosis models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 97–114. [Google Scholar] [CrossRef]
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The Pathobiology of Amyotrophic Lateral Sclerosis: A Proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664. [Google Scholar] [CrossRef]
- Bendotti, C.; Atzori, C.; Piva, R.; Tortarolo, M.; Strong, M.J.; Debiasi, S.; Migheli, A. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol. 2004, 63, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Leigh, P.N.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.E.; Gallo, J.M.; Weller, R.O.; Anderton, B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 1991, 114, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L.; Kim, H.T.; Lee, D.; Collins, G.A. Mechanisms That Activate 26S Proteasomes and Enhance Protein Degradation. Biomolecules 2021, 11, 779. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., 3rd; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Muniyappan, S.; Tran, N.N.; Park, H.; Lee, S.B.; Lee, B.H. Deubiquitination Reactions on the Proteasome for Proteasome Versatility. Int. J. Mol. Sci. 2020, 21, 5312. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Lee, B.H.; Hanna, J.; King, R.W.; Finley, D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol. Cell. Proteomics 2011, 10, R110.003871. [Google Scholar] [CrossRef] [Green Version]
- Koulich, E.; Li, X.; DeMartino, G.N. Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol. Biol. Cell 2008, 19, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.T.; Goldberg, A.L. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J. Biol. Chem. 2017, 292, 9830–9839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aufderheide, A.; Beck, F.; Stengel, F.; Hartwig, M.; Schweitzer, A.; Pfeifer, G.; Goldberg, A.L.; Sakata, E.; Baumeister, W.; Förster, F. Structural characterization of the interaction of Ubp6 with the 26S proteasome. Proc Natl Acad Sci USA 2015, 112, 8626–8631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.L.; Goldberg, A.L. Ubiquitinated proteins promote the association of proteasomes with the deubiquitinating enzyme Usp14 and the ubiquitin ligase Ube3c. Proc. Natl. Acad. Sci. USA 2017, 114, E3404–e3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.H.; Lu, Y.; Prado, M.A.; Shi, Y.; Tian, G.; Sun, S.; Elsasser, S.; Gygi, S.P.; King, R.W.; Finley, D. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 2016, 532, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Muniyappan, S.; Lee, S.-B.; Lee, B.-H. Small-Molecule Inhibitors Targeting Proteasome-Associated Deubiquitinases. Int. J. Mol. Sci. 2021, 22, 6213. [Google Scholar] [CrossRef]
- Lam, Y.A.; Xu, W.; DeMartino, G.N.; Cohen, R.E. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 1997, 385, 737–740. [Google Scholar] [CrossRef]
- Deol, K.K.; Crowe, S.O.; Du, J.; Bisbee, H.; Guenette, R.G.; Strieter, E.R. Proteasome-Bound UCH37 Debranches Ubiquitin Chains to Promote Degradation. Mol. Cell 2020, 80, 796–809. [Google Scholar] [CrossRef]
- Boselli, M.; Lee, B.H.; Robert, J.; Prado, M.A.; Min, S.W.; Cheng, C.; Silva, M.C.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, Y.; Ding, S.; Li, J.; Song, N.; Ren, Y.; Hong, D.; Wu, C.; Li, B.; Wang, F.; et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018, 28, 1186–1194. [Google Scholar] [CrossRef]
- Palmer, A.L.; de Jong, A.; Leestemaker, Y.; Geurink, P.P.; Wijdeven, R.H.; Ovaa, H.; Dolan, B.P. Inhibition of the deubiquitinase Usp14 diminishes direct MHC class I antigen presentation. J. Immunol. 2018, 200, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.J.; Lee, M.J. Dual Function of USP14 Deubiquitinase in Cellular Proteasomal Activity and Autophagic Flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef] [Green Version]
- VerPlank, J.J.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Hu, Y.; Huang, P.; Toleman, C.A.; Paterson, A.J.; Kudlow, J.E. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem. 2007, 282, 22460–22471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, M.; Tsukamoto, O.; Minamino, T.; Asanuma, H.; Fujita, M.; Asano, Y.; Takahama, H.; Sasaki, H.; Higo, S.; Asakura, M.; et al. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J. Mol. Cell. Cardiol. 2009, 46, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; Kukushkin, N.V.; Goldberg, A.L. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E7176–E7185. [Google Scholar] [CrossRef] [Green Version]
- VerPlank, J.J.; Lokireddy, S.; Zhao, J.; Goldberg, A.L. 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc. Natl. Acad. Sci. USA 2019, 116, 4228–4237. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wang, X.; Wang, Z.; Banerjee, S.; Yang, J.; Huang, L.; Dixon, J.E. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 2016, 18, 202–212. [Google Scholar] [CrossRef]
- Djakovic, S.N.; Schwarz, L.A.; Barylko, B.; DeMartino, G.N.; Patrick, G.N. Regulation of the Proteasome by Neuronal Activity and Calcium/Calmodulin-dependent Protein Kinase II*. J. Biol. Chem. 2009, 284, 26655–26665. [Google Scholar] [CrossRef] [Green Version]
- Djakovic, S.N.; Marquez-Lona, E.M.; Jakawich, S.K.; Wright, R.; Chu, C.; Sutton, M.A.; Patrick, G.N. Phosphorylation of Rpt6 Regulates Synaptic Strength in Hippocampal Neurons. J. Neurosci. 2012, 32, 5126. [Google Scholar] [CrossRef] [Green Version]
- Schaler, A.W.; Myeku, N. Cilostazol, a phosphodiesterase 3 inhibitor, activates proteasome-mediated proteolysis and attenuates tauopathy and cognitive decline. Transl. Res. 2018, 193, 31–41. [Google Scholar] [CrossRef]
- Zhu, J.; Mix, E.; Winblad, B. The antidepressant and antiinflammatory effects of rolipram in the central nervous system. CNS Drug Rev. 2001, 7, 387–398. [Google Scholar] [CrossRef]
- ASHP. Cilostazol Monographs for Professionals. Available online: https://www.drugs.com/monograph/cilostazol.html (accessed on 4 August 2021).
- A Trial of Cilostazol in Patients with Mild Cognitive Impairment (COMCID). Available online: https://clinicaltrials.gov/ct2/show/NCT02491268 (accessed on 4 August 2021).
- VerPlank, J.J.S.; Tyrkalska, S.D.; Fleming, A.; Rubinsztein, D.C.; Goldberg, A.L. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 14220. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Wang, Y. p38 MAP kinases in the heart. Gene 2016, 575, 369–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.N.; Yang, Q.Q.; Wang, W.T.; Tian, X.; Feng, F.; Zhang, S.T.; Xia, Y.T.; Wang, J.X.; Zou, Y.W.; Wang, J.Y.; et al. Red nucleus IL-33 facilitates the early development of mononeuropathic pain in male rats by inducing TNF-alpha through activating ERK, p38 MAPK, and JAK2/STAT3. J. Neuroinflammation 2021, 18, 150. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, Y.; Yoon, S.K.; Yoon, J.-B. Osmotic Stress Inhibits Proteasome by p38 MAPK-dependent Phosphorylation*. J. Biol. Chem. 2010, 285, 41280–41289. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atzori, C.; Ghetti, B.; Piva, R.; Srinivasan, A.N.; Zolo, P.; Delisle, M.B.; Mirra, S.S.; Migheli, A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Tortarolo, M.; Veglianese, P.; Calvaresi, N.; Botturi, A.; Rossi, C.; Giorgini, A.; Migheli, A.; Bendotti, C. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol. Cell. Neurosci. 2003, 23, 180–192. [Google Scholar] [CrossRef]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef]
- Huang, Z.-N.; Chen, J.-M.; Huang, L.-C.; Fang, Y.-H.; Her, L.-S. Inhibition of p38 Mitogen–Activated Protein Kinase Ameliorates HAP40 Depletion–Induced Toxicity and Proteasomal Defect in Huntington’s Disease Model. Mol. Neurobiol. 2021, 58, 2704–2723. [Google Scholar] [CrossRef] [PubMed]
- Gaczynska, M.; Rock, K.L.; Spies, T.; Goldberg, A.L. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc. Natl. Acad. Sci. USA 1994, 91, 9213–9217. [Google Scholar] [CrossRef] [Green Version]
- Gaczynska, M.; Goldberg, A.L.; Tanaka, K.; Hendil, K.B.; Rock, K.L. Proteasome subunits X and Y alter peptidase activities in opposite ways to the interferon-gamma-induced subunits LMP2 and LMP7. J. Biol. Chem. 1996, 271, 17275–17280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chondrogianni, N.; Tzavelas, C.; Pemberton, A.J.; Nezis, I.P.; Rivett, A.J.; Gonos, E.S. Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem. 2005, 280, 11840–11850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilchez, D.; Boyer, L.; Morantte, I.; Lutz, M.; Merkwirth, C.; Joyce, D.; Spencer, B.; Page, L.; Masliah, E.; Berggren, W.T.; et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012, 489, 304–308. [Google Scholar] [CrossRef]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw Jennifer, L.; Yamamoto, M.; Kensler Thomas, W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yueh, M.F.; Tukey, R.H. Nrf2-Keap1 signaling pathway regulates human UGT1A1 expression in vitro and in transgenic UGT1 mice. J. Biol. Chem. 2007, 282, 8749–8758. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Chanas, S.A.; Henderson, C.J.; McMahon, M.; Sun, C.; Moffat, G.J.; Wolf, C.R.; Yamamoto, M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem. Soc. Trans. 2000, 28, 33–41. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960. [Google Scholar]
- Jang, J.; Wang, Y.; Kim, H.S.; Lalli, M.A.; Kosik, K.S. Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells 2014, 32, 2616–2625. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.K.; Cho, J.M.; Huang, B.; Shin, S.; Kensler, T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007, 43, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yoshimura, T.; Kumatori, A.; Ichihara, A.; Ikai, A.; Nishigai, M.; Kameyama, K.; Takagi, T. Proteasomes (multi-protease complexes) as 20S ring-shaped particles in a variety of eukaryotic cells. J. Biol. Chem. 1988, 263, 16209–16217. [Google Scholar] [CrossRef]
- Tanaka, K.; Yoshimura, T.; Ichihara, A. Role of substrate in reversible activation of proteasomes (multi-protease complexes) by sodium dodecyl sulfate. J. Biochem. 1989, 106, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ho, P.; Chen, C.H. Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett. 2007, 581, 4955–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsiki, M.; Chondrogianni, N.; Chinou, I.; Rivett, A.J.; Gonos, E.S. The olive constituent oleuropein exhibits proteasome stimulatory properties in vitro and confers life span extension of human embryonic fibroblasts. Rejuvenation Res. 2007, 10, 157–172. [Google Scholar] [CrossRef]
- Tan, Y.; Yu, R.; Pezzuto, J.M. Betulinic Acid-induced Programmed Cell Death in Human Melanoma Cells Involves Mitogen-activated Protein Kinase Activation. Clin. Cancer Res. 2003, 9, 2866–2875. [Google Scholar] [PubMed]
- Ruiz de Mena, I.; Mahillo, E.; Arribas, J.; Castaño, J.G. Kinetic mechanism of activation by cardiolipin (diphosphatidylglycerol) of the rat liver multicatalytic proteinase. Biochem. J. 1993, 296 (Pt 1), 93–97. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Yamada, S. Activation of 20S proteasomes from spinach leaves by fatty acids. Plant Cell Physiol. 1996, 37, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Du, D.-M. Recent Advances in the Synthesis of 2-Imidazolines and Their Applications in Homogeneous Catalysis. Adv. Synth. Catal. 2009, 351, 489–519. [Google Scholar] [CrossRef]
- Mehedi, M.S.A.; Tepe, J.J. Recent Advances in the Synthesis of Imidazolines (2009–2020). Adv. Synth. Catal. 2020, 362, 4189–4225. [Google Scholar] [CrossRef]
- Sharma, V.; Peddibhotla, S.; Tepe, J.J. Sensitization of Cancer Cells to DNA Damaging Agents by Imidazolines. J. Am. Chem. Soc. 2006, 128, 9137–9143. [Google Scholar] [CrossRef]
- Kahlon, D.K.; Lansdell, T.A.; Fisk, J.S.; Hupp, C.D.; Friebe, T.L.; Hovde, S.; Jones, A.D.; Dyer, R.D.; Henry, R.W.; Tepe, J.J. Nuclear Factor-κB Mediated Inhibition of Cytokine Production by Imidazoline Scaffolds. J. Med. Chem. 2009, 52, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, D.K.; Lansdell, T.A.; Fisk, J.S.; Tepe, J.J. Structural–activity relationship study of highly-functionalized imidazolines as potent inhibitors of nuclear transcription factor-κB mediated IL-6 production. Biorg. Med. Chem. 2009, 17, 3093–3103. [Google Scholar] [CrossRef] [PubMed]
- Sztanke, K.; Pasternak, K.; Sidor-Wójtowicz, A.; Truchlińska, J.; Jóźwiak, K. Synthesis of imidazoline and imidazo[2,1-c][1,2,4]triazole aryl derivatives containing the methylthio group as possible antibacterial agents. Biorg. Med. Chem. 2006, 14, 3635–3642. [Google Scholar] [CrossRef]
- Sharma, V.; Lansdell, T.A.; Peddibhotla, S.; Tepe, J.J. Sensitization of Tumor Cells toward Chemotherapy: Enhancing the Efficacy of Camptothecin with Imidazolines. Chem. Biol. 2004, 11, 1689–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, M.D.; Dorgan, R.J.J.; Manger, B.R.; Mamalis, P.; Webster, R.A.B. Isothiourea derivatives of 6-phenyl-2,3,5,6-tetrahydroimidazo[2,1-b]thiazole with broad-spectrum anthelmintic activity. J. Med. Chem. 1987, 30, 1848–1853. [Google Scholar] [CrossRef]
- Crane, L.; Anastassiadou, M.; Hage, S.E.; Stigliani, J.L.; Baziard-Mouysset, G.; Payard, M.; Leger, J.M.; Bizot-Espiard, J.-G.; Ktorza, A.; Caignard, D.-H.; et al. Design and synthesis of novel imidazoline derivatives with potent antihyperglycemic activity in a rat model of type 2 diabetes. Biorg. Med. Chem. 2006, 14, 7419–7433. [Google Scholar] [CrossRef]
- Szabo, B. Imidazoline antihypertensive drugs: A critical review on their mechanism of action. Pharmacol. Ther. 2002, 93, 1–35. [Google Scholar] [CrossRef]
- Savelyeva, M.V.; Baldenkov, G.N.; Kaverina, N.V. Receptor binding potencies of chlorpromazine, trifluoperazine, fluphenazine and their 10-N-substituted analogues. Biomed. Biochim. Acta 1988, 47, 1085–1087. [Google Scholar]
- Medina, D.X.; Caccamo, A.; Oddo, S. Methylene blue reduces aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011, 21, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Flores, J.; Noël, A.; Beauchet, O.; Sjöström, P.J.; LeBlanc, A.C. Methylene blue inhibits Caspase-6 activity, and reverses Caspase-6-induced cognitive impairment and neuroinflammation in aged mice. Acta Neuropathol. Commun. 2019, 7, 210. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-J.; Li, Q.; Liu, D.-L.; Ding, M.-W. Synthesis, fungicidal activity, and sterol 14α-demethylase binding interaction of 2-azolyl-3, 4-dihydroquinazolines on Penicillium digitatum. J. Agric. Food Chem. 2013, 61, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Alphey, M.S.; Jones, D.C.; Shanks, E.J.; Street, I.P.; Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Dihydroquinazolines as a novel class of Trypanosoma brucei trypanothione reductase inhibitors: Discovery, synthesis, and characterization of their binding mode by protein crystallography. J. Med. Chem. 2011, 54, 6514–6530. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, S.J.; Park, S.J.; Lee, M.J.; Rhim, H.; Seo, S.H.; Kim, K.-S. Growth inhibition of human cancer cells in vitro by T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2006, 16, 5014–5017. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Seo, H.N.; Choe, Y.J.; Kim, S.; Oh, C.R.; Kim, Y.D.; Rhim, H.; Choo, D.J.; Kim, J.; Lee, J.Y. T-type Ca2+ channel blockers suppress the growth of human cancer cells. Bioorg. Med. Chem. Lett. 2008, 18, 3899–3901. [Google Scholar] [CrossRef]
- Al-Obaid, A.M.; Abdel-Hamide, S.G.; El-Kashef, H.A.; Alaa, A.-M.; El-Azab, A.S.; Al-Khamees, H.A.; El-Subbagh, H.I. Substituted quinazolines, part 3. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4 (3H)-quinazolinone analogs. Eur. J. Med. Chem. 2009, 44, 2379–2391. [Google Scholar] [CrossRef]
- Jung, S.Y.; Lee, S.H.; Kang, H.B.; Park, H.A.; Chang, S.K.; Kim, J.; Choo, D.J.; Oh, C.R.; Kim, Y.D.; Seo, J.H. Antitumor activity of 3, 4-dihydroquinazoline dihydrochloride in A549 xenograft nude mice. Bioorg. Med. Chem. Lett. 2010, 20, 6633–6636. [Google Scholar] [CrossRef]
- Kang, H.B.; Rim, H.-K.; Park, J.Y.; Choi, H.W.; Choi, D.L.; Seo, J.-H.; Chung, K.-S.; Huh, G.; Kim, J.; Choo, D.J. In vivo evaluation of oral anti-tumoral effect of 3, 4-dihydroquinazoline derivative on solid tumor. Bioorg. Med. Chem. Lett. 2012, 22, 1198–1201. [Google Scholar] [CrossRef]
- Rim, H.-K.; Lee, H.-W.; Choi, I.S.; Park, J.Y.; Choi, H.W.; Choi, J.-H.; Cho, Y.-W.; Lee, J.Y.; Lee, K.-T. T-type Ca2+ channel blocker, KYS05047 induces G1 phase cell cycle arrest by decreasing intracellular Ca2+ levels in human lung adenocarcinoma A549 cells. Bioorg. Med. Chem. Lett. 2012, 22, 7123–7126. [Google Scholar] [CrossRef]
- Jang, S.J.; Choi, H.W.; Choi, D.L.; Cho, S.; Rim, H.-K.; Choi, H.-E.; Kim, K.-S.; Huang, M.; Rhim, H.; Lee, K.-T. In vitro cytotoxicity on human ovarian cancer cells by T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2013, 23, 6656–6662. [Google Scholar] [CrossRef] [PubMed]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marschall, M.; Stamminger, T.; Urban, A.; Wildum, S.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. In Vitro Evaluation of the Activities of the Novel Anticytomegalovirus Compound AIC246 (Letermovir) against Herpesviruses and Other Human Pathogenic Viruses. Antimicrob. Agents Chemother. 2012, 56, 1135–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magyar, C.L.; Wall, T.J.; Davies, S.B.; Campbell, M.V.; Barna, H.A.; Smith, S.R.; Savich, C.J.; Mosey, R.A. Triflic anhydride mediated synthesis of 3,4-dihydroquinazolines: A three-component one-pot tandem procedure. Org. Biomol. Chem. 2019, 17, 7995–8000. [Google Scholar] [CrossRef] [PubMed]
- Vermeiren, C.; de Hemptinne, I.; Vanhoutte, N.; Tilleux, S.; Maloteaux, J.M.; Hermans, E. Loss of metabotropic glutamate receptor-mediated regulation of glutamate transport in chemically activated astrocytes in a rat model of amyotrophic lateral sclerosis. J. Neurochem. 2006, 96, 719–731. [Google Scholar] [CrossRef]
- Benmohamed, R.; Arvanites, A.C.; Kim, J.; Ferrante, R.J.; Silverman, R.B.; Morimoto, R.I.; Kirsch, D.R. Identification of compounds protective against G93A-SOD1 toxicity for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2011, 12, 87–96. [Google Scholar] [CrossRef]
- Santoro, A.M.; Lanza, V.; Bellia, F.; Sbardella, D.; Tundo, G.R.; Cannizzo, A.; Grasso, G.; Arizzi, M.; Nicoletti, V.G.; Alcaro, S.; et al. Pyrazolones Activate the Proteasome by Gating Mechanisms and Protect Neuronal Cells from β-Amyloid Toxicity. ChemMedChem 2020, 15, 302–316. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, D.E.; Tepe, J.J. Advances in Proteasome Enhancement by Small Molecules. Biomolecules 2021, 11, 1789. https://doi.org/10.3390/biom11121789
George DE, Tepe JJ. Advances in Proteasome Enhancement by Small Molecules. Biomolecules. 2021; 11(12):1789. https://doi.org/10.3390/biom11121789
Chicago/Turabian StyleGeorge, Dare E., and Jetze J. Tepe. 2021. "Advances in Proteasome Enhancement by Small Molecules" Biomolecules 11, no. 12: 1789. https://doi.org/10.3390/biom11121789
APA StyleGeorge, D. E., & Tepe, J. J. (2021). Advances in Proteasome Enhancement by Small Molecules. Biomolecules, 11(12), 1789. https://doi.org/10.3390/biom11121789