
Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase?

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Thermodynamic Data
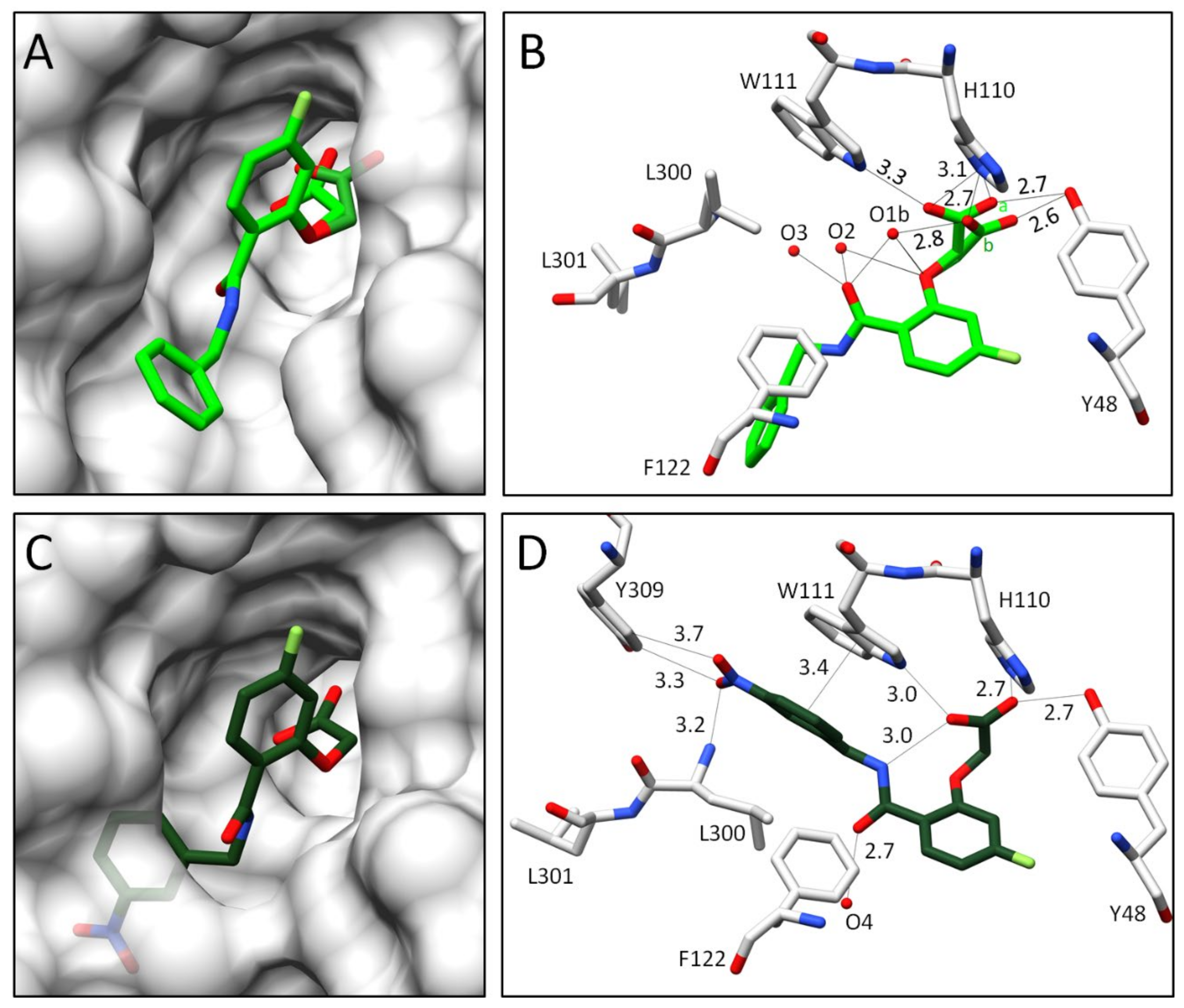
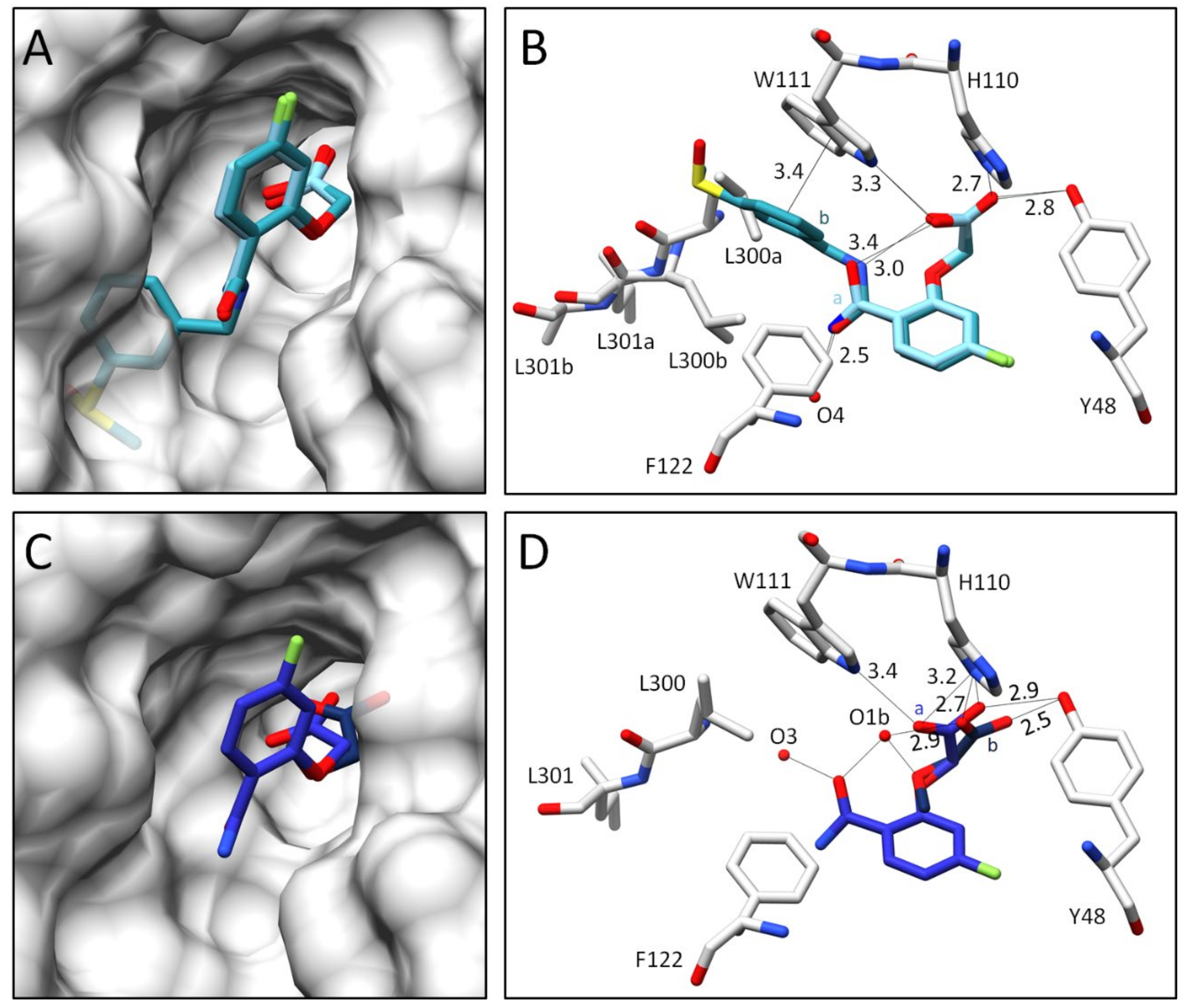
2.2. Crystal Structure Determinations
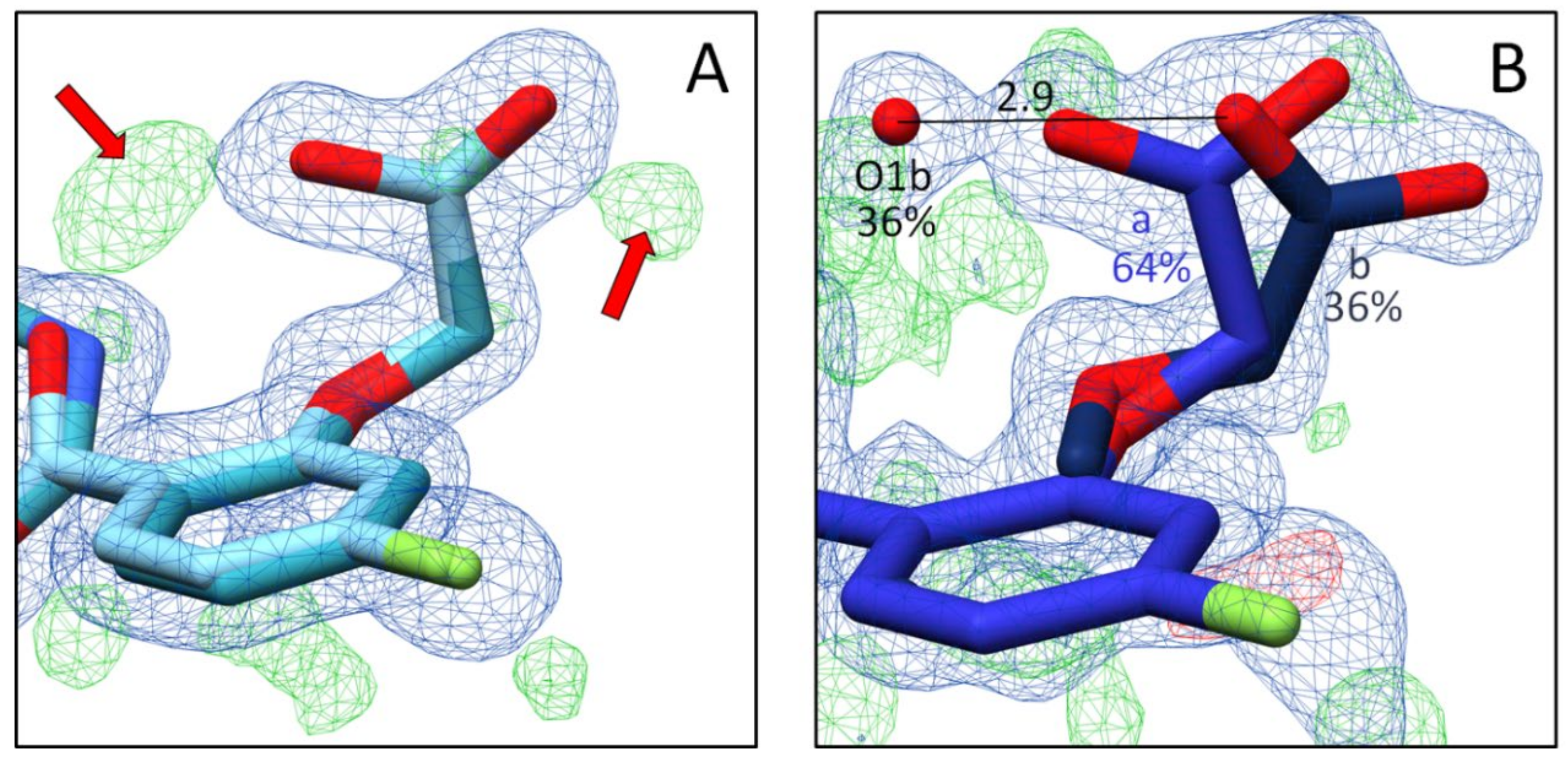
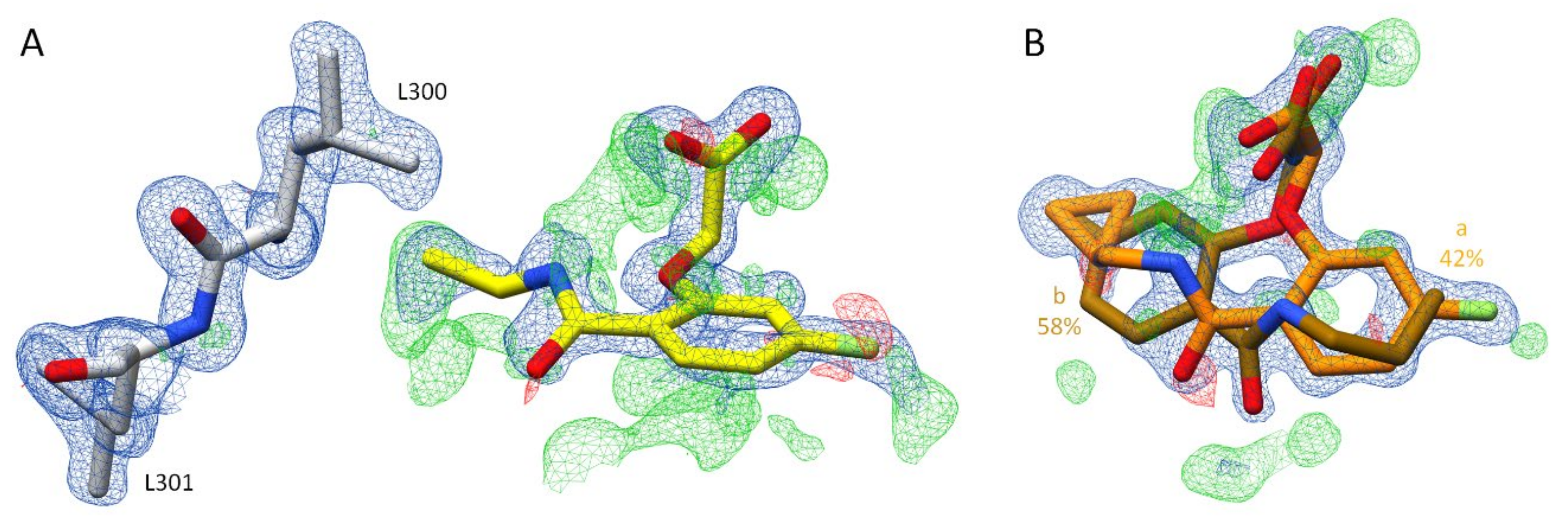
2.2.1. Effects of Different pH Conditions on the Terminal Acetic Acid Carboxy Group at the Parent Scaffold
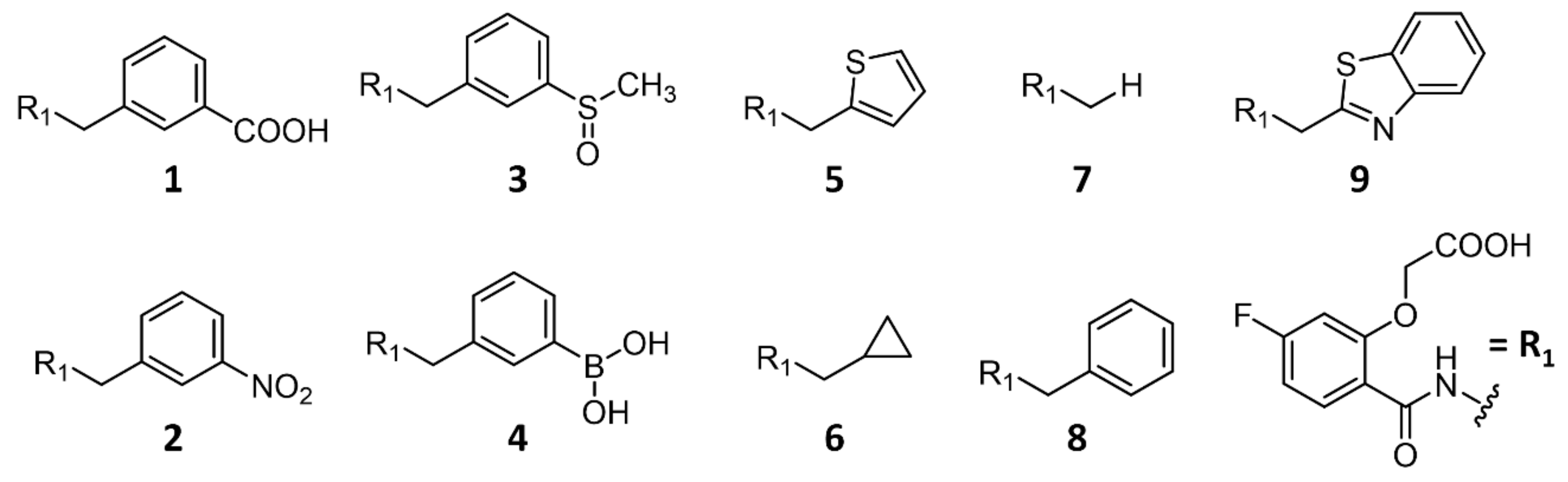
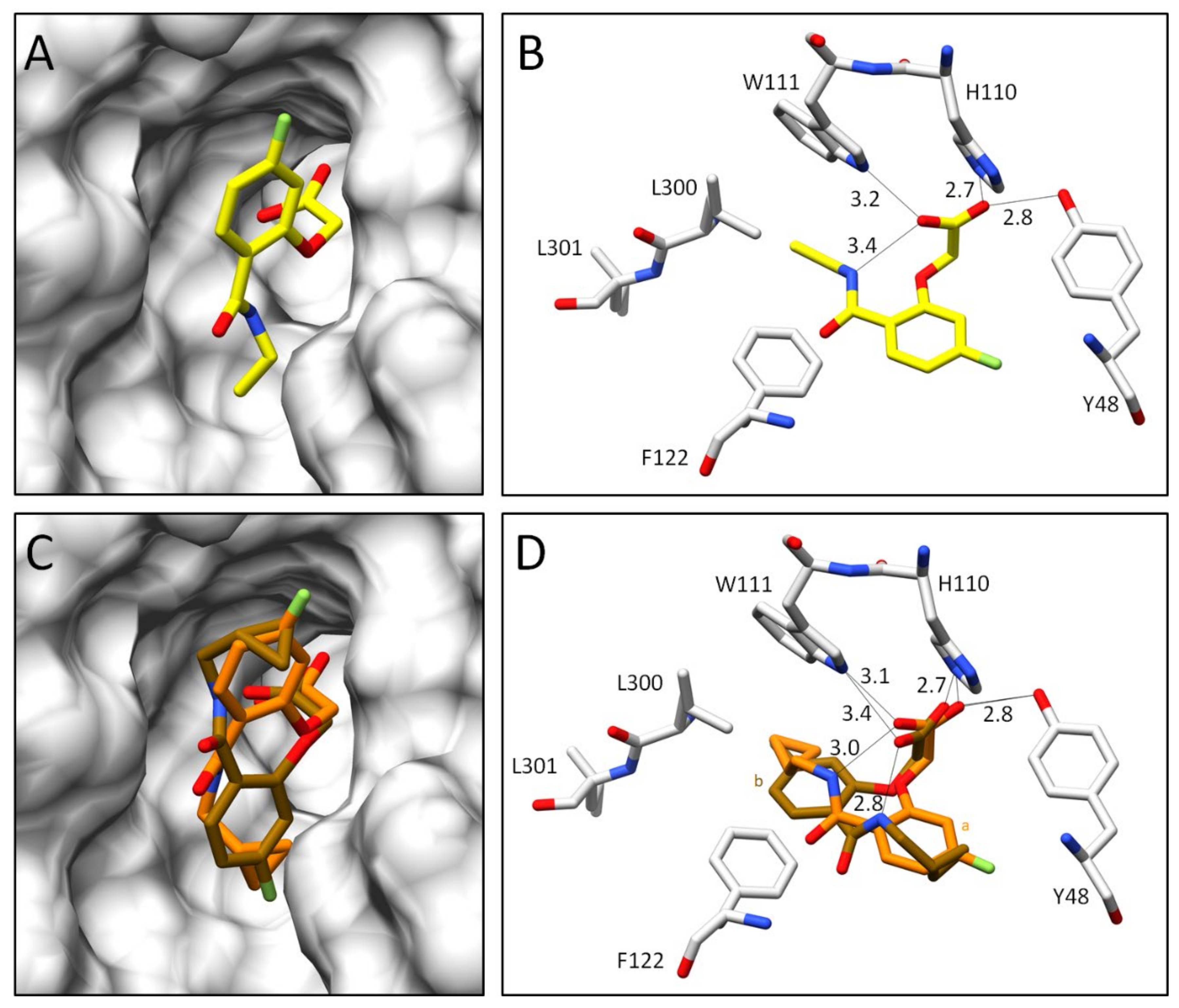
2.2.2. Comparison of the Binding Poses of Inhibitors 1–6
3. Discussion
3.1. Structural and Thermodynamic Comparison
3.2. Comparison of the Electronic Properties of the Terminal Aromatic Substituent
4. Conclusions
5. Materials and Methods
5.1. Protein Expression and Purification
5.2. Isothermal Titration Calorimetry
5.3. Macromolecular Crystallography
5.4. Ab Initio ESP Calculations
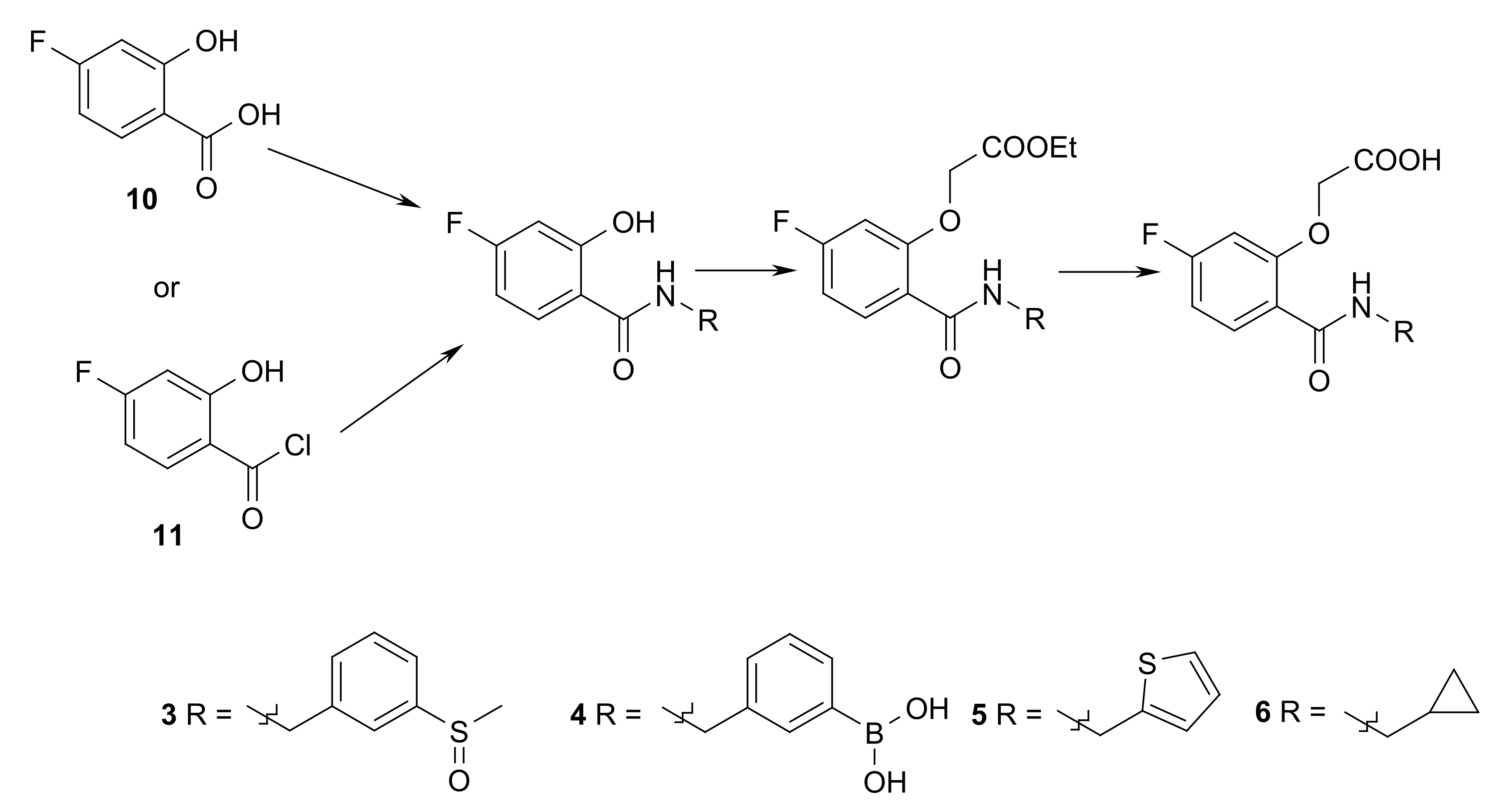
5.5. Ligand Synthesis
5.6. Associated Content
PDB Accession Codes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Å | Ångström (1 Å = 10−10 m) |
| ALR-1 | Aldehyde Reductase |
| ALR-2 | Aldose Reductase |
| B-factor | Debye-Waller factor |
| DAHC | diammonium hydrogen citrate |
| DMSO | Dimethyl sulfoxide |
| DTT | Dithiothreitol |
| EC | Enzyme Commission number |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-hydrochlorid |
| ESI | Electrospray ionization |
| ESP | Electronic Surface Potential area |
| FC | Calculated structure amplitudes |
| FO | Observed structure amplitudes |
| g | Gram |
| G° | Gibbs free energy |
| H° | Enthalpy |
| H-bond | Hydrogen bond |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazine ethane sulfonic acid |
| HOBt | 1-Hydroxybenzotriazol |
| HPLC | High Performance Liquid Chromatography |
| IC50 | Half-maximum inhibitory concentration |
| IPTG | Isopropyl-β-d-thiogalactopyranosid |
| ITC | Isothermal titration calorimetry |
| K | Kelvin |
| k | Kilo |
| Ka | Association constant |
| KD | Dissociation konstant |
| M | Molarity (mol/L) |
| min | Minute |
| MS | Mass spectroscopy |
| NADP+ | Nicotinamide adenine dinucleotide phosphate |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced |
| NMR | Nuclear magnetic resonance |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate buffered saline |
| PEG | Polyethylen glycol |
| pH | Potentialis hydrogenii |
| pKa | Logarithmic acid dissociation constant |
| R-factor | Reliability factor |
| S°SD | EntropyStandard Deviation |
| SDS | Sodium dodecyl sulfate |
| T | Absolute temperature |
| TRIS | Tris(hydroxamethyl)aminomethane |
| X-ray | Röntgen radiation |
References
- Laskowski, R.A.; Luscombe, N.M.; Swindells, M.B.; Thornton, J.M. Protein clefts in molecular recognition and function. Protein Sci. 1996, 5, 2438–2452. [Google Scholar] [PubMed]
- Shah, A.; Shinde, R.; Kare, P.; Hymavathi, V.; Chavan, S.; Elizabeth Sobhia, M. Induced fit binding of aldose reductase inhibitors to AKR1B10. Med. Chem. Res. 2012, 21, 1245–1252. [Google Scholar] [CrossRef]
- Terwesten, F. Proteindynamik Flexibilität in Zielproteinen des Strukturbasierten Wirkstoffdesigns. Ph.D. Thesis, Philipps Universität Marburg, Hessen, Germany, 2017. [Google Scholar]
- Rechlin, C.; Scheer, F.; Terwesten, F.; Wulsdorf, T.; Pol, E.; Fridh, V.; Toth, P.; Diederich, W.E.; Heine, A.; Klebe, G. Price for Opening the Transient Specificity Pocket in Human Aldose Reductase upon Ligand Binding: Structural, Thermodynamic, Kinetic, and Computational Analysis. ACS Chem. Biol. 2017, 12, 1397–1415. [Google Scholar] [CrossRef] [PubMed]
- Acker, T.M.; Gable, J.E.; Bohn, M.-F.; Jaishankar, P.; Thompson, M.C.; Fraser, J.S.; Renslo, A.R.; Craik, C.S. Allosteric Inhibitors, Crystallography, and Comparative Analysis Reveal Network of Coordinated Movement across Human Herpesvirus Proteases. J. Am. Chem. Soc. 2017, 139, 11650–11653. [Google Scholar] [CrossRef]
- Bowman, G.R.; Geissler, P.L. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. USA 2012, 109, 11681–11686. [Google Scholar] [CrossRef]
- Bohren, K.M.; Bullock, B.; Wermuth, B.; Gabbay, K.H. The aldo-keto reductase superfamily. cDNAs and deduced amino acid sequences of human aldehyde and aldose reductases. J. Biol. Chem. 1989, 264, 9547–9561. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Chouthe, R.S.; Sakle, N.S.; Gonjari, I.; Shinde, D.B. Aldose Reductase: A Multi-Disease Target. Curr. Enzym. Inhib. 2014, 10, 2–12. [Google Scholar] [CrossRef]
- Steuber, H.; Heine, A.; Podjarny, A.; Klebe, G. Merging the Binding Sites of Aldose and Aldehyde Reductase for Detection of Inhibitor Selectivity-determining Features. J. Mol. Biol. 2008, 379, 991–1016. [Google Scholar] [CrossRef]
- Van Zandt, M.C.; Sibley, E.O.; McCann, E.E.; Combs, K.J.; Flam, B.; Sawicki, D.R.; Sabetta, A.; Carrington, A.; Sredy, J.; Howard, E.; et al. Design and synthesis of highly potent and selective (2-arylcarbamoyl- phenoxy)-acetic acid inhibitors of aldose reductase for treatment of chronic diabetic complications. Bioorganic Med. Chem. 2004, 12, 5661–5675. [Google Scholar] [CrossRef]
- Krimmer, S.G.; Klebe, G. Thermodynamics of protein-ligand interactions as a reference for computational analysis: How to assess accuracy, reliability and relevance of experimental data. J. Comput. Aided. Mol. Des. 2015, 29, 867–883. [Google Scholar] [CrossRef]
- Broecker, J.; Vargas, C.; Keller, S. Revisiting the optimal c-value for isothermal titration calorimetry. Anal. Biochem. 2011, 418, 307–309. [Google Scholar] [CrossRef]
- Tellinghuisen, J. Isothermal titration calorimetry at very low c. Anal. Biochem. 2008, 373, 395–397. [Google Scholar] [CrossRef]
- Turnbull, W.B.; Daranas, A.H. On the Value of c: Can Low Affinity Systems Be Studied by Isothermal Titration Calorimetry? J. Am. Chem. Soc. 2003, 125, 14859–14866. [Google Scholar] [CrossRef]
- Steuber, H.; Czodrowski, P.; Sotriffer, C.A.; Klebe, G. Tracing Changes in Protonation: A Prerequisite to Factorize Thermodynamic Data of Inhibitor Binding to Aldose Reductase. J. Mol. Biol. 2007, 373, 1305–1320. [Google Scholar] [CrossRef]
- Steuber, H.; Heine, A.; Klebe, G. Structural and Thermodynamic Study on Aldose Reductase: Nitro-substituted Inhibitors with Strong Enthalpic Binding Contribution. J. Mol. Biol. 2007, 368, 618–638. [Google Scholar] [CrossRef]
- Krämer, O. Theoretische und Experimentelle Untersuchungen zur Inhibition des Enzyms Aldose Reduktase. Ph.D. Thesis, Philipps Universität Marburg, Hessen, Germany, 2003. [Google Scholar]
- Misuri, L.; Cappiello, M.; Balestri, F.; Moschini, R.; Barracco, V.; Mura, U.; Del-Corso, A. The use of dimethylsulfoxide as a solvent in enzyme inhibition studies: The case of aldose reductase. J. Enzyme Inhib. Med. Chem. 2017, 32, 1152–1158. [Google Scholar] [CrossRef]
- Ruiz, F.; Mitschler, A.; Joachimiak, A.; Karplus, M.; Podjarny, A. The crystallographic structure of the aldose reductase—IDD552 complex shows direct proton donation from tyrosine 48 research papers. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, D60, 1347–1354. [Google Scholar] [CrossRef]
- Rechlin, C. Insights into Protein-Ligand Molecular Recognition: Thermodynamic, Kinetic and Structural Characterization of Inhibitor Binding to Aldose Reductase and Carbonic Anhydrase II. Ph.D. Thesis, Philipps Universität Marburg, Hessen, Germany, 2015. [Google Scholar]
- Kheirjou, S.; Abedin, A.; Fattahi, A. Theoretical descriptors response to the calculations of the relative pKa values of some boronic acids in aqueous solution: A DFT study. Comput. Theor. Chem. 2012, 1000, 1–5. [Google Scholar] [CrossRef]
- Jezuita, A.; Ejsmont, K.; Szatylowicz, H. Substituent effects of nitro group in cyclic compounds. Struct. Chem. 2021, 32, 179–203. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Lamour, V.; Barth, P.; Rogniaux, H.; Poterszman, A.; Howard, E.; Mitschler, A.; Van Dorsselaer, A.; Podjarny, A.; Moras, D. Production of Crystals of Human Aldose Reductase with Very High Resolution Diffraction. Acta Crystallogr. Sect. D Struct. Biol. 1999, 55, 721–723. [Google Scholar] [CrossRef]
- Kabsch, W. Scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computational Project. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. Sect. D 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Adams, P.D.; Pavel, V.; Chen, V.B.; Ian, W.; Echols, N.; Moriarty, N.W.; Read, R.J.; Richardson, D.C.; Jane, S.; Thomas, C. PHENIX: A comprehensive Python-based system for macromolecular structure solution research papers. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics research papers. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Schönemann, W.; Cramer, J.; Mühlethaler, T.; Fiege, B.; Silbermann, M.; Rabbani, S.; Dätwyler, P.; Zihlmann, P.; Jakob, R.P.; Sager, C.P.; et al. Improvement of Aglycone π-Stacking Yields Nanomolar to Sub-nanomolar FimH Antagonists. Chem. Med. Chem. 2019, 14, 749–757. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef]
- Ngo, K. Synthese von D-Phe und D-DiPhe-Pro-Basierenden Inhibitoren zur Thermodynamischen Untersuchung der Bindungseigenschaften in der S1-Tasche von Thrombin. Ph.D. Thesis, Philipps Universität Marburg, Hessen, Germany, 2016. [Google Scholar]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | KD [µM] | ΔG° [kJ/mol] | ΔH° [kJ/mol] | −TΔS° [kJ/mol] | Titration Mode |
|---|---|---|---|---|---|
| 1 | 36.2 ± 11.4 (a) | −25.4 ± 0.7 (a) | −36.4 ± 2.9 (a) | 11.0 ± 3.6 (a) | displ. (a,b) |
| 2 | 0.018 ± 0.003 (a) | −44.3 ± 0.5 (a) | −65.6 ± 0.5 (a) | 21.3 ± 0.9 (a) | displ. (a,c) |
| 3 | 5.0 ± 0.6 | −30.3 ± 0.3 | −39.1 ± 0.9 | 8.8 ± 0.9 | direct |
| 4 | 2.3 ± 0.3 | −32.2 ± 0.3 | −45.0 ± 0.7 | 12.8 ± 0.8 | direct |
| 5 | 77.6 ± 15.9 | −23.4 ± 0.5 | −30.0 ± 1.4 | 6.6 ± 1.9 | displ. (b) |
| 6 | 107.7 ± 16.6 | −22.6 ± 0.4 | −37.5 ± 0.8 | 14.9 ± 1.2 | displ. (b) |
| 7 | 6.6 ± 0.4 (a) | −29.6 ± 0.2 (a) | −35.4 ± 0.3 (a) | 5.8 ± 0.4 (a) | direct (a) |
| 7 | 7.2 ± 0.9 (a) | −29.4 ± 0.3 (a) | −39.0 ± 1.0 (a) | 9.6 ± 1.2 (a) | displ. (a,b) |
| 8 | 7.5 ± 1.4 (a) | −29.3 ± 0.4 (a) | −29.9 ± 0.4 (a) | 0.6 ± 0.4 (a) | direct (a) |
| 8 | 9.1 ± 0.7 (a) | −28.8 ± 0.2 (a) | −30.6 ± 0.5 (a) | 1.8 ± 0.7 (a) | displ. (a,b) |
| Inhibitor | KD [µM] | ΔG° [kJ/mol] | ΔH° [kJ/mol] | –TΔS° [kJ/mol] | Titration Mode |
|---|---|---|---|---|---|
| 9 (from this study) | 0.42 ± 0.08 | −36.4 ± 0.5 | −53.3 ± 0.5 | 16.9 ± 0.7 | direct |
| 9 (derived from [4]) | 0.05 ± 0.01 | −41.5 ± 0.4 | −54.0 ± 1.0 | 12.5 ± 1.1 | direct |
| Inhibitor | PDB Code | Resolution [Å] | Pocket State |
|---|---|---|---|
| 1 | 4YS1 (a) | 1.07 | closed |
| 2 | 4QBX (a) | 0.98 | open |
| 3 | 6TUF | 1.15 | hybrid |
| 4 | 6TUC | 1.06 | closed |
| 5 | not deposited | 0.93 | closed |
| 6 | 6SWY | 0.93 | closed |
| 7 | 4PUU (a) | 1.14 | closed |
| 8 | 4Q7B (a) | 1.19 | open |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandner, A.; Ngo, K.; Sager, C.P.; Scheer, F.; Daude, M.; Diederich, W.E.; Heine, A.; Klebe, G. Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase? Biomolecules 2021, 11, 1837. https://doi.org/10.3390/biom11121837
Sandner A, Ngo K, Sager CP, Scheer F, Daude M, Diederich WE, Heine A, Klebe G. Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase? Biomolecules. 2021; 11(12):1837. https://doi.org/10.3390/biom11121837
Chicago/Turabian StyleSandner, Anna, Khang Ngo, Christoph P. Sager, Frithjof Scheer, Michael Daude, Wibke E. Diederich, Andreas Heine, and Gerhard Klebe. 2021. "Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase?" Biomolecules 11, no. 12: 1837. https://doi.org/10.3390/biom11121837
APA StyleSandner, A., Ngo, K., Sager, C. P., Scheer, F., Daude, M., Diederich, W. E., Heine, A., & Klebe, G. (2021). Which Properties Allow Ligands to Open and Bind to the Transient Binding Pocket of Human Aldose Reductase? Biomolecules, 11(12), 1837. https://doi.org/10.3390/biom11121837