
A Deep Convolutional Neural Network for Prediction of Peptide Collision Cross Sections in Ion Mobility Spectrometry



Abstract
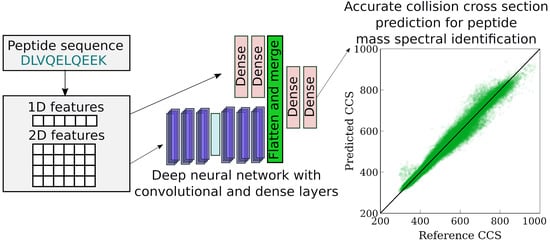
:
1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. Input Features for the Deep Neural Network
2.3. Neural Network Architecture and Training
3. Results and Discussion
3.1. Accuracy of Collision Cross Section Prediction
3.2. Comparison with Previous Works
3.3. Model Averaging for Further Improvement of Accuracy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedev, A.T.; Damoc, E.; Makarov, A.A.; Samgina, T.Y. Discrimination of Leucine and Isoleucine in Peptides Sequencing with Orbitrap Fusion Mass Spectrometer. Anal. Chem. 2014, 86, 7017–7022. [Google Scholar] [CrossRef] [PubMed]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Kanu, A.B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H.H. Ion Mobility-Mass Spectrometry. J. Mass Spectrom. 2008, 43, 1–22. [Google Scholar] [CrossRef]
- Valentine, S.J.; Ewing, M.A.; Dilger, J.M.; Glover, M.S.; Geromanos, S.; Hughes, C.; Clemmer, D.E. Using Ion Mobility Data to Improve Peptide Identification: Intrinsic Amino Acid Size Parameters. J. Proteome Res. 2011, 10, 2318–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, E.S.; Livesay, E.A.; Orton, D.J.; Moore, R.J.; Danielson, W.F.; Prior, D.C.; Ibrahim, Y.M.; LaMarche, B.L.; Mayampurath, A.M.; Schepmoes, A.A.; et al. An LC-IMS-MS Platform Providing Increased Dynamic Range for High-Throughput Proteomic Studies. J. Proteome Res. 2010, 9, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Prianichnikov, N.; Koch, H.; Koch, S.; Lubeck, M.; Heilig, R.; Brehmer, S.; Fischer, R.; Cox, J. MaxQuant Software for Ion Mobility Enhanced Shotgun Proteomics. Mol. Cell. Proteom. 2020, 19, 1058–1069. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Ren, Y.; Yang, J.; Ren, Z.; Yang, H.; Liu, S. Improved Peptide Retention Time Prediction in Liquid Chromatography through Deep Learning. Anal. Chem. 2018, 90, 10881–10888. [Google Scholar] [CrossRef]
- Bouwmeester, R.; Gabriels, R.; Hulstaert, N.; Martens, L.; Degroeve, S. DeepLC Can Predict Retention Times for Peptides That Carry As-yet Unseen Modifications. Nat. Methods 2021, 18, 1363–1369. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In Silico Spectral Libraries by Deep Learning Facilitate Data-Independent Acquisition Proteomics. Nat. Commun. 2020, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdrakhimov, D.A.; Bubis, J.A.; Gorshkov, V.; Kjeldsen, F.; Gorshkov, M.V.; Ivanov, M.V. Biosaur: An Open-source Python Software for Liquid Chromatography–Mass Spectrometry Peptide Feature Detection with Ion Mobility Support. Rapid Commun. Mass Spectrom. 2021, e9045. [Google Scholar] [CrossRef]
- Ivanov, M.V.; Bubis, J.A.; Gorshkov, V.; Abdrakhimov, D.A.; Kjeldsen, F.; Gorshkov, M.V. Boosting MS1-Only Proteomics with Machine Learning Allows 2000 Protein Identifications in Single-Shot Human Proteome Analysis Using 5 Min HPLC Gradient. J. Proteome Res. 2021, 20, 1864–1873. [Google Scholar] [CrossRef]
- Shah, A.R.; Agarwal, K.; Baker, E.S.; Singhal, M.; Mayampurath, A.M.; Ibrahim, Y.M.; Kangas, L.J.; Monroe, M.E.; Zhao, R.; Belov, M.E.; et al. Machine Learning Based Prediction for Peptide Drift Times in Ion Mobility Spectrometry. Bioinformatics 2010, 26, 1601–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Yeung, D.; Spicer, V.; Ogata, K.; Krokhin, O.; Ishihama, Y. Sequence-Specific Model for Predicting Peptide Collision Cross Section Values in Proteomic Ion Mobility Spectrometry. J. Proteome Res. 2021, 20, 3600–3610. [Google Scholar] [CrossRef] [PubMed]
- Mosier, P.D.; Counterman, A.E.; Jurs, P.C.; Clemmer, D.E. Prediction of Peptide Ion Collision Cross Sections from Topological Molecular Structure and Amino Acid Parameters. Anal. Chem. 2002, 74, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Köhler, N.D.; Brunner, A.-D.; Wanka, J.-M.H.; Voytik, E.; Strauss, M.T.; Theis, F.J.; Mann, M. Deep Learning the Collisional Cross Sections of the Peptide Universe from a Million Experimental Values. Nat. Commun. 2021, 12, 1185. [Google Scholar] [CrossRef]
- Meyer, J.G. Deep Learning Neural Network Tools for Proteomics. Cell Rep. Methods 2021, 1, 100003. [Google Scholar] [CrossRef]
- Plante, P.-L.; Francovic-Fontaine, É.; May, J.C.; McLean, J.A.; Baker, E.S.; Laviolette, F.; Marchand, M.; Corbeil, J. Predicting Ion Mobility Collision Cross-Sections Using a Deep Neural Network: DeepCCS. Anal. Chem. 2019, 91, 5191–5199. [Google Scholar] [CrossRef]
- Matyushin, D.D.; Buryak, A.K. Gas Chromatographic Retention Index Prediction Using Multimodal Machine Learning. IEEE Access 2020, 8, 223140–223155. [Google Scholar] [CrossRef]
- Matyushin, D.D.; Sholokhova, A.Y.; Buryak, A.K. Deep Learning Based Prediction of Gas Chromatographic Retention Indices for a Wide Variety of Polar and Mid-Polar Liquid Stationary Phases. Int. J. Mol. Sci. 2021, 22, 9194. [Google Scholar] [CrossRef]
- Barley, M.H.; Turner, N.J.; Goodacre, R. Improved Descriptors for the Quantitative Structure–Activity Relationship Modeling of Peptides and Proteins. J. Chem. Inf. Model. 2018, 58, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Long, H.; Bo, Z.; Wang, Y.; Wu, Y. New Descriptors of Amino Acids and Their Application to Peptide QSAR Study. Peptides 2008, 29, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, R.; Martens, L.; Degroeve, S. Generalized Calibration Across Liquid Chromatography Setups for Generic Prediction of Small-Molecule Retention Times. Anal. Chem. 2020, 92, 6571–6578. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Description of Features |
|---|---|
| 0–20 | One hot encoded amino acid (including oxidized methionine as the 21st amino acid). One of these features is set as 1 and others are set as 0. |
| 21–25 | Elemental composition of the residue: integer values that describe a number of each element (H, C, N, O, S). |
| 26–31 | Six binary features that show whether the side-chain of the amino acid is acidic (D, E), modified (non-standard), has an amide group (N, Q), is non-polar (G, A, V, L, I, P, M, F, V), is small (P, G, A, S), is uncharged polar (S, T, N, Q, C, Y, oxidized methionine residue). |
| 32–35 | Four binary features that show whether the side-chain of the amino acid is aliphatic non-polar (V, I, L, G, A), is aromatic (W, F, Y), is positively charged (K, R, H), has a hydroxyl group (S, T, Y). |
| 36 | For N, D (Asp, Asn) this feature is 1, 0 elsewhere. |
| 37 | For E, Q (Glu, Gln) this feature is 1, 0 elsewhere. |
| 38–43 | Descriptors of amino acids from references [21,22], six float values, constant for each amino acid type. |
| 44 | Always 0 for all amino acids, 1 for padding symbol. |
| Data Set | Training Iterations | Features | Accuracy Measures | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RMSE, Å2 | MAE, Å2 | MPE,% | MdPE, % | Δ90, % | R2 | r | |||
| Training set | 14,160 | Full | 12.6 | 7.5 | 1.46 | 1.00 | 3.21 | 0.987 | 0.993 |
| 26,000 | Full | 11.7 | 7.1 | 1.40 | 0.96 | 3.05 | 0.988 | 0.994 | |
| 76,110 | Full | 9.8 | 6.2 | 1.23 | 0.87 | 2.68 | 0.992 | 0.996 | |
| 30,000 | Reduced | 11.4 | 7.0 | 1.38 | 0.95 | 3.03 | 0.989 | 0.994 | |
| Test set | 14,160 | Full | 13.1 | 7.7 | 1.50 | 1.02 | 3.28 | 0.985 | 0.993 |
| 26,000 | Full | 12.8 | 7.6 | 1.47 | 1.00 | 3.25 | 0.986 | 0.993 | |
| 76,110 | Full | 13.2 | 7.6 | 1.48 | 0.99 | 3.22 | 0.985 | 0.993 | |
| 30,000 | Reduced | 13.3 | 7.8 | 1.52 | 1.03 | 3.32 | 0.985 | 0.992 | |
| ProteomeTools test set | 14,160 | Full | 14.9 | 9.4 | 1.84 | 1.28 | 4.02 | 0.983 | 0.991 |
| 26,000 | Full | 14.6 | 9.3 | 1.82 | 1.28 | 3.95 | 0.983 | 0.992 | |
| 76,110 | Full | 15.3 | 9.5 | 1.86 | 1.27 | 4.06 | 0.982 | 0.991 | |
| 30,000 | Reduced | 15.4 | 9.7 | 1.90 | 1.32 | 4.17 | 0.982 | 0.991 | |
| Subset | This Work | Previous Results [16] | ||||
|---|---|---|---|---|---|---|
| MdPE, % | Δ90, % | r | MdPE, % | Δ90, % | r | |
| Full ProteomeTools test set | 1.28 | 3.95 | 0.992 | 1.40 | 4.0 | 0.992 |
| Charge state + 2 | 1.06 | 3.06 | 0.985 | 1.2 | ||
| Charge state + 3 | 1.77 | 5.49 | 0.938 | 1.8 | ||
| Charge state + 4 | 1.95 | 5.47 | 0.886 | 2.0 | ||
| CCS values 0–400 | 1.06 | 3.02 | 0.920 | 1.2 | ||
| CCS values 400–800 | 1.38 | 4.32 | 0.986 | 1.5 | ||
| CCS values 800–1200 | 2.30 | 5.71 | 0.695 | 2.2 | ||
| Subset | This Work | |||||
|---|---|---|---|---|---|---|
| RMSE, Å2 | MAE, Å2 | MdPE, % | Δ90, % | r | R2 | |
| Human plasma | 26.7 | 20.4 | 3.85 | 7.99 | 0.989 | 0.979 |
| Mouse plasma | 26.9 | 21.7 | 4.53 | 8.55 | 0.990 | 0.980 |
| Shewanella oneidensis | 17.2 | 11.8 | 1.85 | 5.18 | 0.988 | 0.976 |
| Subset | This Work | Previous Results [13] | ||
|---|---|---|---|---|
| R2 | MSE, s2 | R2 | MSE, s2 | |
| Charge state + 2 | 0.9634 | 0.287 | 0.9620 | 0.290 |
| Charge state + 3 | 0.9179 | 0.715 | 0.9062 | 0.813 |
| Charge state + 4 | 0.9266 | 0.705 | 0.9308 | 0.556 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samukhina, Y.V.; Matyushin, D.D.; Grinevich, O.I.; Buryak, A.K. A Deep Convolutional Neural Network for Prediction of Peptide Collision Cross Sections in Ion Mobility Spectrometry. Biomolecules 2021, 11, 1904. https://doi.org/10.3390/biom11121904
Samukhina YV, Matyushin DD, Grinevich OI, Buryak AK. A Deep Convolutional Neural Network for Prediction of Peptide Collision Cross Sections in Ion Mobility Spectrometry. Biomolecules. 2021; 11(12):1904. https://doi.org/10.3390/biom11121904
Chicago/Turabian StyleSamukhina, Yulia V., Dmitriy D. Matyushin, Oksana I. Grinevich, and Aleksey K. Buryak. 2021. "A Deep Convolutional Neural Network for Prediction of Peptide Collision Cross Sections in Ion Mobility Spectrometry" Biomolecules 11, no. 12: 1904. https://doi.org/10.3390/biom11121904