
The Cross Marks the Spot: The Emerging Role of JmjC Domain-Containing Proteins in Myeloid Malignancies


Abstract
:1. Introduction
2. Results
2.1. The KDM2 Family
2.1.1. KDM2A
2.1.2. KDM2B
2.2. The KDM3 Family
2.2.1. KDM3A
2.2.2. KDM3B
2.2.3. KDM3C
2.3. The KDM4 Family
2.3.1. KDM4A
2.3.2. KDM4B
2.3.3. KDM4C
2.3.4. KDM4D
2.4. The KDM5 Family
2.4.1. KDM5A
2.4.2. KDM5B
2.5. The KDM6 Family
2.5.1. KDM6A
2.5.2. KDM6B
2.6. The KDM7 Family
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Mosammaparast, N.; Shi, Y. Reversal of Histone Methylation: Biochemical and Molecular Mechanisms of Histone Demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Øra, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-Specific Demethylase 1 Is Strongly Expressed in Poorly Differentiated Neuroblastoma: Implications for Therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zang, J.; Whetstine, J.; Hong, X.; Davrazou, F.; Kutateladze, T.; Simpson, M.; Mao, Q.; Pan, C.-H.; Dai, S.; et al. Structural Insights into Histone Demethylation by JMJD2 Family Members. Cell 2006, 125, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Franci, G.; Ciotta, A.; Altucci, L. The Jumonji family: Past, present and future of histone demethylases in cancer. Biomol. Concepts 2014, 5, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Markolovic, S.; Leissing, T.M.; Chowdhury, R.; E Wilkins, S.; Lu, X.; Schofield, C.J. Structure–function relationships of human JmjC oxygenases—demethylases versus hydroxylases. Curr. Opin. Struct. Biol. 2016, 41, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, Q.; Wong, S.H.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Tan, L.; Nagata, Y.; Takemura, T.; Asahina, A.; Yokota, D.; Yagyu, T.; Shibata, K.; Fujisawa, S.; Ohnishi, K. JmjC-domain containing histone demethylase 1B-mediated p15 Ink4b suppression promotes the proliferation of leukemic progenitor cells through modulation of cell cycle progression in acute myeloid leukemia. Mol. Carcinog. 2011, 52, 57–69. [Google Scholar] [CrossRef]
- He, J.; Nguyen, A.T.; Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 2011, 117, 3869–3880. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Hirabayashi, Y. Hematopoietic neoplastic diseases develop in C3H/He and C57BL/6 mice after benzene exposure: Strain differences in bone marrow tissue responses observed using microarrays. Chem. Interact. 2010, 184, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Guardavaccaro, D.; Kuchay, S.M.; Kato, H.; Poleshko, A.; Basrur, V.; Elenitoba-Johnson, K.; Katz, R.A.; Pagano, M. KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 2008, 7, 3539–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, D.P.; Aplan, P.D. Chromosomal Rearrangements Leading to MLL Gene Fusions: Clinical and Biological Aspects. Cancer Res. 2008, 68, 10024–10027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boom, V.V.D.; Maat, H.; Geugien, M.; López, A.R.; Sotoca, A.M.; Jaques, J.; Brouwers-Vos, A.Z.; Fusetti, F.; Groen, R.; Yuan, H.; et al. Non-canonical PRC1.1 Targets Active Genes Independent of H3K27me3 and Is Essential for Leukemogenesis. Cell Rep. 2016, 14, 332–346. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Nagamachi, A.; Takubo, K.; Yamasaki, N.; Matsui, H.; Kanai, A.; Nakata, Y.; Ikeda, K.; Konuma, T.; Oda, H.; et al. Fbxl10 overexpression in murine hematopoietic stem cells induces leukemia involving metabolic activation and upregulation of Nsg2. Blood 2015, 125, 3437–3446. [Google Scholar] [CrossRef] [Green Version]
- Karoopongse, E.; Yeung, C.; Byon, J.; Ramakrishnan, A.; Holman, Z.J.; Jiang, P.Y.Z.; Yu, Q.; Deeg, H.J.; Marcondes, A.M. The KDM2B- Let-7b -EZH2 Axis in Myelodysplastic Syndromes as a Target for Combined Epigenetic Therapy. PLoS ONE 2014, 9, e107817. [Google Scholar] [CrossRef]
- Andricovich, J.; Kai, Y.; Peng, W.; Foudi, A.; Tzatsos, A. Histone demethylase KDM2B regulates lineage commitment in normal and malignant hematopoiesis. J. Clin. Investig. 2016, 126, 905–920. [Google Scholar] [CrossRef]
- Jafek, J.L.; Shakya, A.; Tai, P.-Y.; Ibarra, A.; Kim, H.; Maddox, J.; Chumley, J.; Spangrude, G.J.; Miles, R.R.; Kelley, T.W.; et al. Transcription factor Oct1 protects against hematopoietic stress and promotes acute myeloid leukemia. Exp. Hematol. 2019, 76, 38–48.e2. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Xu, C.; Jiang, G.; Wang, H.; Zhang, J. KDM3B suppresses APL progression by restricting chromatin accessibility and facilitating the ATRA-mediated degradation of PML/RARα. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Nagel, S.; Quentmeier, H.; Wang, Z.; Pommerenke, C.; Dirks, W.G.; MacLeod, R.A.F.; Drexler, H.G.; Hu, Z. KDM3B shows tumor-suppressive activity and transcriptionally regulates HOXA1 through retinoic acid response elements in acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Gomes, I.; Horrigan, S.K.; Kravarusic, J.; Mar, B.; Arbieva, Z.; Chyna, B.; Fulton, N.C.; Edassery, S.; Raza, A.; et al. A novel nuclear protein, 5qNCA (LOC51780) is a candidate for the myeloid leukemia tumor suppressor gene on chromosome 5 band q31. Oncogene 2001, 20, 6946–6954. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, X.; Zhang, X.; Wang, Y.; Wang, X.; Hu, L.; Zhao, Y.; Wang, H.; Wang, Z.; Wang, H.; et al. Modulators of histone demethylase JMJD1C selectively target leukemic stem cells. FEBS Open Bio 2021, 11, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Schimek, V.; Björn, N.; Pellé, L.; Svedberg, A.; Gréen, H. JMJD1C knockdown affects myeloid cell lines proliferation, viability, and gemcitabine/carboplatin-sensitivity. Pharmacogenet. Genom. 2021, 31, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Staehle, H.F.; Heinemann, J.; Gruender, A.; Omlor, A.M.; Pahl, H.L.; Jutzi, J.S. Jmjd1c is dispensable for healthy adult hematopoiesis and Jak2V617F-driven myeloproliferative disease initiation in mice. PLoS ONE 2020, 15, e0228362. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, L.; Hu, L.; Dirks, W.; Zhao, Y.; Wei, Z.; Chen, D.; Li, Z.; Wang, Z.; Han, Y.; et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int. J. Cancer 2020, 146, 400–412. [Google Scholar] [CrossRef]
- Izaguirre-Carbonell, J.; Christiansen, L.; Burns, R.; Schmitz, J.; Li, C.; Mokry, R.L.; Bluemn, T.; Zheng, Y.; Shen, J.; Carlson, K.-S.; et al. Critical role of Jumonji domain of JMJD1C in MLL-rearranged leukemia. Blood Adv. 2019, 3, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Salik, B.; Connerty, P.; Vick, B.; Leung, H.; Pijning, A.; Jeremias, I.; Spiekermann, K.; Trahair, T.; Liu, T.; et al. JMJD1C-mediated metabolic dysregulation contributes to HOXA9-dependent leukemogenesis. Leukemia 2019, 33, 1400–1410. [Google Scholar] [CrossRef]
- Peeken, J.C.; Jutzi, J.S.; Wehrle, J.; Koellerer, C.; Staehle, H.F.; Becker, H.; Schoenwandt, E.; Seeger, T.S.; Schanne, D.H.; Gothwal, M.; et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood 2018, 131, 2065–2073. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Chen, M.; Eng, R.; DeJong, J.; Sinha, A.U.; Rahnamay, N.F.; Koche, R.; Al-Shahrour, F.; Minehart, J.; Chen, C.-W.; et al. MLL-AF9– and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J. Clin. Investig. 2016, 126, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhu, N.; Liu, X.; Laurent, B.; Tang, Z.; Eng, R.; Shi, Y.; Armstrong, S.A.; Roeder, R.G. JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors. Genes Dev. 2015, 29, 2123–2139. [Google Scholar] [CrossRef] [Green Version]
- Sroczynska, P.; Cruickshank, V.A.; Bukowski, J.-P.; Miyagi, S.; Bagger, F.O.; Walfridsson, J.; Schuster, M.B.; Porse, B.; Helin, K. shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood 2014, 123, 1870–1882. [Google Scholar] [CrossRef]
- Agger, K.; Miyagi, S.; Pedersen, M.T.; Kooistra, S.M.; Johansen, J.V.; Helin, K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016, 30, 1278–1288. [Google Scholar] [CrossRef] [Green Version]
- Massett, M.E.; Monaghan, L.; Patterson, S.; Mannion, N.; Bunschoten, R.P.; Hoose, A.; Marmiroli, S.; Liskamp, R.M.J.; Jørgensen, H.G.; Vetrie, D.; et al. A KDM4A-PAF1-mediated epigenomic network is essential for acute myeloid leukemia cell self-renewal and survival. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Chaudhary, K.; Deb, S.; Moniaux, N.; Ponnusamy, M.P.; Batra, S.K. Human RNA polymerase II-associated factor complex: Dysregulation in cancer. Oncogene 2007, 26, 7499–7507. [Google Scholar] [CrossRef] [PubMed]
- Milan, T.; Celton, M.; Lagacé, K.; Roques, É.; Safa-Tahar-Henni, S.; Bresson, E.; Bergeron, A.; Hebert, J.; Meshinchi, S.; Cellot, S.; et al. Epigenetic changes in human model KMT2A leukemias highlight early events during leukemogenesis. Haematologica 2020. [Google Scholar] [CrossRef]
- Xue, L.; Li, C.; Ren, J.; Wang, Y. KDM4C contributes to cytarabine resistance in acute myeloid leukemia via regulating the miR-328-3p/CCND2 axis through MALAT1. Ther. Adv. Chronic Dis. 2021, 12, 2040622321997259. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Wang, P.; Han, G.; Zhang, T.; Chang, J.; Yin, R.; Shan, Y.; Wen, J.; Xie, X.; et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 2020, 27, 81–97.e8. [Google Scholar] [CrossRef]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W.E. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.S.; Zhou, J.; Cheong, L.-L.; Liu, S.-C.; Qian, J.; Guo, T.; Sze, S.K.; Zeng, Q.; Chng, W.J. LEO1 Is Regulated by PRL-3 and Mediates Its Oncogenic Properties in Acute Myelogenous Leukemia. Cancer Res. 2014, 74, 3043–3053. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Cao, X.; Mo, L. Overexpression of KDM4D promotes acute myeloid leukemia cell development by activating MCL-1. Am. J. Transl. Res. 2021, 13, 2308–2319. [Google Scholar]
- E De Rooij, J.D.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; Van Galen, J.F.; Beverloo, H.B.; Baruchel, A.; Trka, J.; Reinhardt, D.; Sonneveld, E.; Zimmermann, M.B.; et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013, 27, 2280–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Zeng, J.; Wang, X.; Wang, X.; Huang, T.; Fu, Y.; Sun, T.; Jia, J.; Chen, C. Histone demethylase RBP2 decreases miR-21 in blast crisis of chronic myeloid leukemia. Oncotarget 2014, 6, 1249–1261. [Google Scholar] [CrossRef]
- Zhou, M.; Yin, X.; Zheng, L.; Fu, Y.; Wang, Y.; Cui, Z.; Gao, Z.; Wang, X.; Huang, T.; Jia, J.; et al. miR-181d/RBP2/NF-κB p65 Feedback Regulation Promotes Chronic Myeloid Leukemia Blast Crisis. Front. Oncol. 2021, 11, 654411. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Lam, Y.M.; He, Z.; Zheng, Y.; Li, L.; Zhang, Y.; Li, C.; Mbadhi, M.N.; Zheng, L.; Cheng, Z.; et al. Histone lysine demethylase KDM5B maintains chronic myeloid leukemia via multiple epigenetic actions. Exp. Hematol. 2020, 82, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Goode, D.L.; Iwasaki, M.; Wei, M.C.; Kuo, H.-P.; Zhu, L.; Schneidawind, D.; Duque-Afonso, J.; Weng, Z.; Cleary, M.L. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015, 28, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Orgueira, A.M.; Raíndo, A.P.; López, M.C.; Arias, J.Á.D.; Pérez, M.S.G.; Rodríguez, B.A.; Vence, N.A.; Pérez, L.B.; Ferro, R.F.; Ferreiro, M.A.; et al. Personalized Survival Prediction of Patients With Acute Myeloblastic Leukemia Using Gene Expression Profiling. Front. Oncol. 2021, 11, 657191. [Google Scholar] [CrossRef]
- Stewart, M.H.; Albert, M.; Sroczynska, P.; Cruickshank, V.A.; Guo, Y.; Rossi, D.J.; Helin, K.; Enver, T. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal in mice. Blood 2015, 125, 2075–2078. [Google Scholar] [CrossRef] [Green Version]
- Cellot, S.; Hope, K.J.; Chagraoui, J.; Sauvageau, M.; Deneault, É.; MacRae, T.; Mayotte, N.; Wilhelm, B.T.; Landry, J.R.; Ting, S.B.; et al. RNAi screen identifies Jarid1b as a major regulator of mouse HSC activity. Blood 2013, 122, 1545–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, P.-F.X.P.-F.; Tao, Y.-F.T.Y.-F.; Hu, S.-Y.H.S.-Y.; Cao, L.C.L.; Lu, J.L.J.; Wang, J.W.J.; Feng, X.F.X.; Pan, J.P.J.; Chai, Y.-H.C.Y.-H. mRNA expression profiling of histone modifying enzymes in pediatric acute monoblastic leukemia. Die Pharm. Int. J. Pharm. Sci. 2017, 72, 177–186. [Google Scholar]
- Zhan, D.; Zhang, Y.; Xiao, P.; Zheng, X.; Ruan, M.; Zhang, J.; Chen, A.; Zou, Y.; Chen, Y.; Huang, G.; et al. Whole exome sequencing identifies novel mutations of epigenetic regulators in chemorefractory pediatric acute myeloid leukemia. Leuk. Res. 2018, 65, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xu, L.; Xu, Q.; Yu, L.; Zhao, D.; Chen, P.; Wang, W.; Wang, Y.; Han, G.; Chen, C.D. Utx loss causes myeloid transformation. Leukemia 2018, 32, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Thieme, S.; Gyárfás, T.; Richter, C.; Özhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 2013, 121, 2462–2473. [Google Scholar] [CrossRef] [Green Version]
- Gozdecka, M.; Meduri, E.; Mazan, M.; Tzelepis, K.; Dudek, M.; Knights, A.J.; Pardo, M.; Yu, L.; Choudhary, J.; Metzakopian, E.; et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018, 50, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Stief, S.M.; Hanneforth, A.-L.; Weser, S.; Mattes, R.; Carlet, M.; Liu, W.-H.; Bartoschek, M.; Moreno, H.D.; Oettle, M.; Kempf, J.; et al. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia 2020, 34, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, L.; Zhu, Y.; Xu, R.; Deng, Z.; Liu, X.; Ding, Y.; Wang, C.; Shi, Y.; Bei, L.; et al. KDM6A promotes imatinib resistance through YY1-mediated transcriptional upregulation of TRKA independently of its demethylase activity in chronic myelogenous leukemia. Theranostics 2021, 11, 2691–2705. [Google Scholar] [CrossRef]
- Liu, J.; Mercher, T.; Scholl, C.; Brumme, K.; Gilliland, D.G.; Zhu, N. A functional role for the histone demethylase UTX in normal and malignant hematopoietic cells. Exp. Hematol. 2012, 40, 487–498.e3. [Google Scholar] [CrossRef]
- Biswas, M.; Chatterjee, S.S.; Boila, L.D.; Chakraborty, S.; Banerjee, D.; Sengupta, A. MBD3/NuRD loss participates with KDM6A program to promoteDOCK5/8expression and Rac GTPase activation in human acute myeloid leukemia. FASEB J. 2019, 33, 5268–5286. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, R.; Dimicoli, S.; Bueso-Ramos, C.; Neuberg, D.; Pierce, S.; Wang, H.; Yang, H.; Jia, Y.; Zheng, H.; et al. Global H3K4me3 genome mapping reveals alterations of innate immunity signaling and overexpression of JMJD3 in human myelodysplastic syndrome CD34+ cells. Leukemia 2013, 27, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, M.; Sheng, M.; Zhang, P.; Chen, Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.-C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J. Cancer Res. Clin. Oncol. 2018, 144, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zheng, H.; Bao, N.; Jiang, S.; Bueso-Ramos, C.E.; Khoury, J.; Class, C.; Lu, Y.; Lin, K.; Yang, H.; et al. KDM6B overexpression activates innate immune signaling and impairs hematopoiesis in mice. Blood Adv. 2018, 2, 2491–2504. [Google Scholar] [CrossRef]
- Mallaney, C.; Ostrander, E.L.; Celik, H.; Kramer, A.C.; Martens, A.; Kothari, A.; Koh, W.K.; Haussler, E.; Iwamori, N.; Gontarz, P.; et al. Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 2019, 33, 2506–2521. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ye, Y.; Wang, X.; Lu, B.; Guo, Z.; Wu, S. JMJD3-regulated expression of IL-6 is involved in the proliferation and chemosensitivity of acute myeloid leukemia cells. Biol. Chem. 2019, 402, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-H.; Zhu, K.-Y.; Chen, J.; Liu, X.-Z.; Xu, P.-F.; Zhang, W.; Yan, L.; Guo, H.-Z.; Zhu, J. JMJD3 facilitates C/EBPβ-centered transcriptional program to exert oncorepressor activity in AML. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Maciejewski, J.P. The functional mechanisms of mutations in myelodysplastic syndrome. Leukemia 2019, 33, 2779–2794. [Google Scholar] [CrossRef]
- Stomper, J.; Meier, R.; Ma, T.; Pfeifer, D.; Ihorst, G.; Blagitko-Dorfs, N.; Greve, G.; Zimmer, D.; Platzbecker, U.; Hagemeijer, A.; et al. Integrative study of EZH2 mutational status, copy number, protein expression and H3K27 trimethylation in AML/MDS patients. Clin. Epigenet. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.; Liu, C.L.; Bratman, S.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Hock, H. A complex Polycomb issue: The two faces of EZH2 in cancer. Genes Dev. 2012, 26, 751–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaeirad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Scholl, C.; Bansal, D.; Döhner, K.; Eiwen, K.; Huntly, B.; Lee, B.H.; Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Döhner, H.; et al. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J. Clin. Investig. 2007, 117, 1037–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, V.P.S.; Cusan, M.; Deshpande, A.; Hiddemann, W.; Quintanilla-Martinez, L.; Humphries, R.K.; Bohlander, S.; Feuring-Buske, M.; Buske, C. Ectopic expression of the homeobox gene Cdx2 is the transforming event in a mouse model of t(12;13)(p13;q12) acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 817–822. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, R.N.; Kannourakis, G.; Wall, M.; Campbell, L.J. A cryptic deletion in 5q31.2 provides further evidence for a minimally deleted region in myelodysplastic syndromes. Cancer Genet. 2011, 204, 187–194. [Google Scholar] [CrossRef]
- Wang, K.; Wang, P.; Shi, J.; Zhu, X.; He, M.; Jia, X.; Yang, X.; Qiu, F.; Jin, W.; Qian, M.; et al. PML/RARα Targets Promoter Regions Containing PU.1 Consensus and RARE Half Sites in Acute Promyelocytic Leukemia. Cancer Cell 2010, 17, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, M.; Jin, W.; Zhu, X.; Jia, X.; Yang, X.; Du, Y.; Wang, K.; Zhang, J. Structurally differentiated cis-elements that interact with PU.1 are functionally distinguishable in acute promyelocytic leukemia. J. Hematol. Oncol. 2013, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Schwemmers, S.; Hexner, E.O.; Pahl, H.L. AML1 is overexpressed in patients with myeloproliferative neoplasms and mediates JAK2V617F-independent overexpression of NF-E2. Blood 2010, 116, 254–266. [Google Scholar] [CrossRef]
- Goerttler, P.S.; Kreutz, C.; Donauer, J.; Faller, D.; Maiwald, T.; Marz, E.; Rumberger, B.; Sparna, T.; Schmitt-Graff, A.; Wilpert, J.; et al. Gene expression profiling in polycythaemia vera: Overexpression of transcription factor NF-E2. Br. J. Haematol. 2005, 129, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.B.; Gründer, A.; Hadlich, T.; Wehrle, J.; Gothwal, M.; Bogeska, R.; Seeger, T.S.; Kayser, S.; Pham, K.-B.; Jutzi, J.S.; et al. A novel murine model of myeloproliferative disorders generated by overexpression of the transcription factor NF-E2. J. Exp. Med. 2012, 209, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Gründer, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Basu, T.; Pellmann, M.; Kaiser, S.; Steinemann, D.; Sanders, M.A.; Hinai, A.S.A.; Zeilemaker, A.; Kovacs, S.B.; Koellerer, C.; et al. Altered NFE2 activity predisposes to leukemic transformation and myelosarcoma with AML-specific aberrations. Blood 2019, 133, 1766–1777. [Google Scholar] [CrossRef]
- Pedersen, M.T.; Kooistra, S.M.; Radzisheuskaya, A.; Laugesen, A.; Johansen, J.V.; Hayward, D.G.; Nilsson, J.; Agger, K.; Helin, K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016, 35, 1550–1564. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, M.T.; Agger, K.; Laugesen, A.; Johansen, J.V.; Cloos, P.A.C.; Christensen, J.; Helin, K. The Demethylase JMJD2C Localizes to H3K4me3-Positive Transcription Start Sites and Is Dispensable for Embryonic Development. Mol. Cell. Biol. 2014, 34, 1031–1045. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Nishimura, K.; Miyagi, S.; Messling, J.-E.; Rasmussen, K.D.; Helin, K. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 2019, 134, 1154–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagger, F.O.; Kinalis, S.; Rapin, N. BloodSpot: A database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019, 47, D881–D885. [Google Scholar] [CrossRef] [Green Version]
- Broughton, S.; Dhagat, U.; Hercus, T.; Nero, T.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The GM-CSF/IL-3/IL-5 cytokine receptor family: From ligand recognition to initiation of signaling. Immunol. Rev. 2012, 250, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark. Res. 2014, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Serio, J.; Ropa, J.; Chen, W.; Mysliwski, M.; Saha, N.; Chen, L.; Wang, J.; Miao, H.; Cierpicki, T.; Grembecka, J.; et al. The PAF complex regulation of Prmt5 facilitates the progression and maintenance of MLL fusion leukemia. Oncogene 2018, 37, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.; Hess, J.L. The PAF Complex Synergizes with MLL Fusion Proteins at HOX Loci to Promote Leukemogenesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Souto, J.A.; Sarno, F.; Nebbioso, A.; Papulino, C.; Álvarez, R.; Lombino, J.; Perricone, U.; Padova, A.; Altucci, L.; De Lera, Á.R. A New Family of Jumonji C Domain-Containing KDM Inhibitors Inspired by Natural Product Purpurogallin. Front. Chem. 2020, 8, 312. [Google Scholar] [CrossRef]
- Laouedj, M.; Tardif, M.R.; Gil, L.; Raquil, M.-A.; Lachhab, A.; Pelletier, M.; Tessier, P.A.; Barabé, F.; Lachaab, A. S100A9 induces differentiation of acute myeloid leukemia cells through TLR4. Blood 2017, 129, 1980–1990. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.; Cao, Y.-X.; Luo, Z.-Y.; Liao, P.; Lu, Z.-W. LncRNA MALAT1 promotes cell proliferation and imatinib resistance by sponging miR-328 in chronic myelogenous leukemia. Biochem. Biophys. Res. Commun. 2018, 507, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Zhou, H.; Yao, W.; Meng, L.; Lang, B. lncRNA TUG1 Promotes Cisplatin Resistance by Regulating CCND2 via Epigenetically Silencing miR-194-5p in Bladder Cancer. Mol. Ther. Nucleic Acids 2019, 16, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Bi, C.; Chng, W.-J.; Cheong, L.-L.; Liu, S.-C.; Mahara, S.; Tay, K.-G.; Zeng, Q.; Li, J.; Guo, K.; et al. PRL-3, a Metastasis Associated Tyrosine Phosphatase, Is Involved in FLT3-ITD Signaling and Implicated in Anti-AML Therapy. PLoS ONE 2011, 6, e19798. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Yang, L.; Xie, M.; Lin, C.; Merkurjev, D.; Yang, J.C.; Tanasa, B.; Oh, S.; Zhang, J.; Ohgi, K.A.; et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA 2014, 111, 9235–9240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinohara, K.; Wu, H.-J.; Vigneau, S.; McDonald, T.O.; Igarashi, K.J.; Yamamoto, K.N.; Madsen, T.; Fassl, A.; Egri, S.B.; Papanastasiou, M.; et al. KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell 2018, 34, 939–953.e9. [Google Scholar] [CrossRef] [Green Version]
- Kirtana, R.; Manna, S.; Patra, S.K. Molecular mechanisms of KDM5A in cellular functions: Facets during development and disease. Exp. Cell Res. 2020, 396, 112314. [Google Scholar] [CrossRef] [PubMed]
- van Zutven, L.J.C.M.; Önen, E.; Velthuizen, S.C.J.M.; van Drunen, E.; von Bergh, A.R.M.; Heuvel-Eibrink, M.M.V.D.; Veronese, A.; Mecucci, C.; Negrini, M.; de Greef, G.E.; et al. Identification ofNUP98 abnormalities in acute leukemia:JARID1A (12p13) as a new partner gene. Genes Chromosom. Cancer 2006, 45, 437–446. [Google Scholar] [CrossRef]
- Choi, H.-J.; Joo, H.-S.; Won, H.-Y.; Min, K.-W.; Kim, H.-Y.; Son, T.; Oh, Y.-H.; Lee, J.-Y.; Kong, G. Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. J. Natl. Cancer Inst. 2017, 110, 400–410. [Google Scholar] [CrossRef]
- Cardin, S.; Bilodeau, M.; Roussy, M.; Aubert, L.; Milan, T.; Jouan, L.; Rouette, A.; Laramée, L.; Gendron, P.; Duchaine, J.; et al. Human models of NUP98-KDM5A megakaryocytic leukemia in mice contribute to uncovering new biomarkers and therapeutic vulnerabilities. Blood Adv. 2019, 3, 3307–3321. [Google Scholar] [CrossRef]
- Hara, Y.; Shiba, N.; Yamato, G.; Ohki, K.; Tabuchi, K.; Sotomatsu, M.; Tomizawa, D.; Kinoshita, A.; Arakawa, H.; Saito, A.M.; et al. Patients aged less than 3 years with acute myeloid leukaemia characterize a molecularly and clinically distinct subgroup. Br. J. Haematol. 2020, 188, 528–539. [Google Scholar] [CrossRef]
- Hara, Y.; Shiba, N.; Ohki, K.; Tabuchi, K.; Yamato, G.; Park, M.-J.; Tomizawa, D.; Kinoshita, A.; Shimada, A.; Arakawa, H.; et al. Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosom. Cancer 2017, 56, 394–404. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.D.E.; Branstetter, C.; Ma, J.; Li, Y.; Walsh, M.P.; Cheng, J.; Obulkasim, A.; Dang, J.; Easton, J.; Verboon, L.J.; et al. Pediatric non–Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 2017, 49, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.; Zhou, M.-M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. NUP98–PHF23 Is a Chromatin-Modifying Oncoprotein That Causes a Wide Array of Leukemias Sensitive to Inhibition of PHD Histone Reader Function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, T.B.; Uluisik, R.C.; Van Linden, A.A.; Jones, K.L.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. Increased HDAC Activity and c-MYC Expression Mediate Acquired Resistance to WEE1 Inhibition in Acute Leukemia. Front. Oncol. 2020, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Wang, Y.; Lang, Y.; Zhang, Y. KDM5A promotes proliferation and EMT in ovarian cancer and closely correlates with PTX resistance. Mol. Med. Rep. 2017, 16, 3573–3580. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Ge, Z.; Wang, L.; Li, Q.; Wang, N.; Björkholm, M.; Jia, J.; Xu, D. The Histone Demethylase RBP2 Is Overexpressed in Gastric Cancer and Its Inhibition Triggers Senescence of Cancer Cells. Gastroenterology 2010, 138, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Wu, J.; Dombkowski, A.; Zhang, K.; Holowatyj, A.; Boerner, J.L.; Yang, Z.-Q. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res. 2012, 4, 247–256. [Google Scholar]
- Teng, Y.-C.; Lee, C.-F.; Li, Y.-S.; Chen, Y.-R.; Hsiao, P.-W.; Chan, M.-Y.; Lin, F.-M.; Huang, H.-D.; Chen, Y.-T.; Jeng, Y.-M.; et al. Histone Demethylase RBP2 Promotes Lung Tumorigenesis and Cancer Metastasis. Cancer Res. 2013, 73, 4711–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; A Tindell, C.; Wilson, C.; E Williamson, K.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banelli, B.; Carra, E.; Barbieri, F.; Würth, R.; Parodi, F.; Pattarozzi, A.; Carosio, R.; Forlani, A.; Allemanni, G.; Marubbi, D.; et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 2015, 14, 3418–3429. [Google Scholar] [CrossRef] [Green Version]
- He, Z.-K.; Xue, S.; Zhang, Y.-H.; Li, L.; Xia, Y.-J.; Wang, X.; Shi, X.; Liu, Y.; Xu, Z.; Li, C.; et al. Expression Levels of JARID1B, Hes1 and MMP-9 Genes in CML Patients Treated with Imatinib Mesylate. Europe 2019, 7, 1071–1076. [Google Scholar]
- Xhabija, B.; Kidder, B.L. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin. Cancer Biol. 2019, 57, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Montano, M.M.; Yeh, I.-J.; Chen, Y.; Hernandez, C.; Kiselar, J.G.; De La Fuente, M.; Lawes, A.M.; Nieman, M.T.; Kiser, P.D.; Jacobberger, J.; et al. Inhibition of the histone demethylase, KDM5B, directly induces re-expression of tumor suppressor protein HEXIM1 in cancer cells. Breast Cancer Res. 2019, 21, 1–15. [Google Scholar] [CrossRef]
- Pospisil, V.; Vargova, K.S.; Kokavec, J.; Rybarova, J.; Savvulidi, F.G.; Jonasova, A.; Nečas, E.; Zavadil, J.; Laslo, P.; Stopka, T. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. EMBO J. 2011, 30, 4450–4464. [Google Scholar] [CrossRef]
- Petruk, S.; Mariani, S.A.; De Dominici, M.; Porazzi, P.; Minieri, V.; Cai, J.; Iacovitti, L.; Flomenberg, N.; Calabretta, B.; Mazo, A. Structure of Nascent Chromatin Is Essential for Hematopoietic Lineage Specification. Cell Rep. 2017, 19, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Fujishima, N.; Kohmaru, J.; Koyota, S.; Kuba, K.; Saga, T.; Omokawa, A.; Moritoki, Y.; Ueki, S.; Ishida, F.; Nakao, S.; et al. Clonal hematopoiesis in adult pure red cell aplasia. Sci. Rep. 2021, 11, 1–7. [Google Scholar] [CrossRef]
- Bandara, W.M.S.; Rathnayake, A.I.S.; Neththikumara, N.F.; Goonasekera, H.W.; Dissanayake, V.H. Comparative Analysis of the Genetic Variants in Haematopoietic Stem/Progenitor and Mesenchymal Stem Cell Compartments in de novo Myelodysplastic Syndromes. Blood Cells Mol. Dis. 2021, 88, 102535. [Google Scholar] [CrossRef] [PubMed]
- Greif, P.A.; Hartmann, L.; Vosberg, S.; Stief, S.M.; Mattes, R.; Hellmann, I.; Metzeler, K.; Herold, T.; Bamopoulos, S.; Kerbs, P.; et al. Evolution of Cytogenetically Normal Acute Myeloid Leukemia During Therapy and Relapse: An Exome Sequencing Study of 50 Patients. Clin. Cancer Res. 2018, 24, 1716–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowska, A.M.; Makishima, H.; Tiu, R.V.; Szpurka, H.; Huang, Y.; Traina, F.; Visconte, V.; Sugimoto, Y.; Prince, C.; O’Keefe, C.; et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 2011, 118, 3932–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartman, L.D.; Larson, D.E.; Xiang, Z.; Ding, L.; Chen, K.; Lin, L.; Cahan, P.; Klco, J.M.; Welch, J.S.; Li, C.; et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J. Clin. Investig. 2011, 121, 1445–1455. [Google Scholar] [CrossRef] [Green Version]
- Foulds, C.E.; Nelson, M.L.; Blaszczak, A.G.; Graves, B.J. Ras/Mitogen-Activated Protein Kinase Signaling Activates Ets-1 and Ets-2 by CBP/p300 Recruitment. Mol. Cell. Biol. 2004, 24, 10954–10964. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Pan, X.; Chen, X.; Chen, M.; Shi, K.; Xu, J.; Zheng, J.; Niu, T.; Chen, C.; Shuai, X.; et al. Epigenetic drug library screening identified an LSD1 inhibitor to target UTX-deficient cells for differentiation therapy. Signal Transduct. Target. Ther. 2019, 4, 11. [Google Scholar] [CrossRef]
- Boila, L.D.; Chatterjee, S.S.; Banerjee, D.; Sengupta, A. KDM6 and KDM4 histone lysine demethylases emerge as molecular therapeutic targets in human acute myeloid leukemia. Exp. Hematol. 2018, 58, 44–51.e7. [Google Scholar] [CrossRef]
- Torcia, M.; Braccilaudiero, L.; Lucibello, M.; Nencioni, L.; Labardi, D.; Rubartelli, A.; Cozzolino, F.; Aloe, L.; Garaci, E. Nerve Growth Factor Is an Autocrine Survival Factor for Memory B Lymphocytes. Cell 1996, 85, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Davare, M.A.; Druker, B.J.; Tognon, C.E. Revisiting NTRKs as an emerging oncogene in hematological malignancies. Leukemia 2019, 33, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Dubanet, L.; Bentayeb, H.; Petit, B.; Olivrie, A.; Saada, S.; de la Cruz-Morcillo, M.A.; Lalloué, F.; Gourin, M.-P.; Bordessoule, D.; Faumont, N.; et al. Anti-apoptotic role and clinical relevance of neurotrophins in diffuse large B-cell lymphomas. Br. J. Cancer 2015, 113, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Beutel, G.; Rhein, M.; Meyer, J.; Koenecke, C.; Neumann, T.; Yang, M.; Krauter, J.; von Neuhoff, N.; Heuser, M.; et al. High-affinity neurotrophin receptors and ligands promote leukemogenesis. Blood 2009, 113, 2028–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, A.; Arnett, J.; Dunn, S.; Williams, D.A.; Cancelas, J.A. Rac2 GTPase deficiency depletes BCR-ABL+ leukemic stem cells and progenitors in vivo. Blood 2010, 116, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, H.; Uruno, T.; Shirai, T.; Takaya, D.; Matsunaga, S.; Setoyama, D.; Watanabe, M.; Kukimoto-Niino, M.; Oisaki, K.; Ushijima, M.; et al. Targeting Ras-Driven Cancer Cell Survival and Invasion through Selective Inhibition of DOCK1. Cell Rep. 2017, 19, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.K.; Cancelas, J.A.; Chae, H.-D.; Cox, A.D.; Keller, P.J.; Perrotti, D.; Neviani, P.; Druker, B.J.; Setchell, K.D.; Zheng, Y.; et al. Rac Guanosine Triphosphatases Represent Integrating Molecular Therapeutic Targets for BCR-ABL-Induced Myeloproliferative Disease. Cancer Cell 2007, 12, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.C.; Fennell, K.A.; Chan, Y.-C.; Rambow, F.; Yeung, M.M.; Vassiliadis, D.; Lara, L.; Yeh, P.; Martelotto, L.G.; Rogiers, A.; et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]

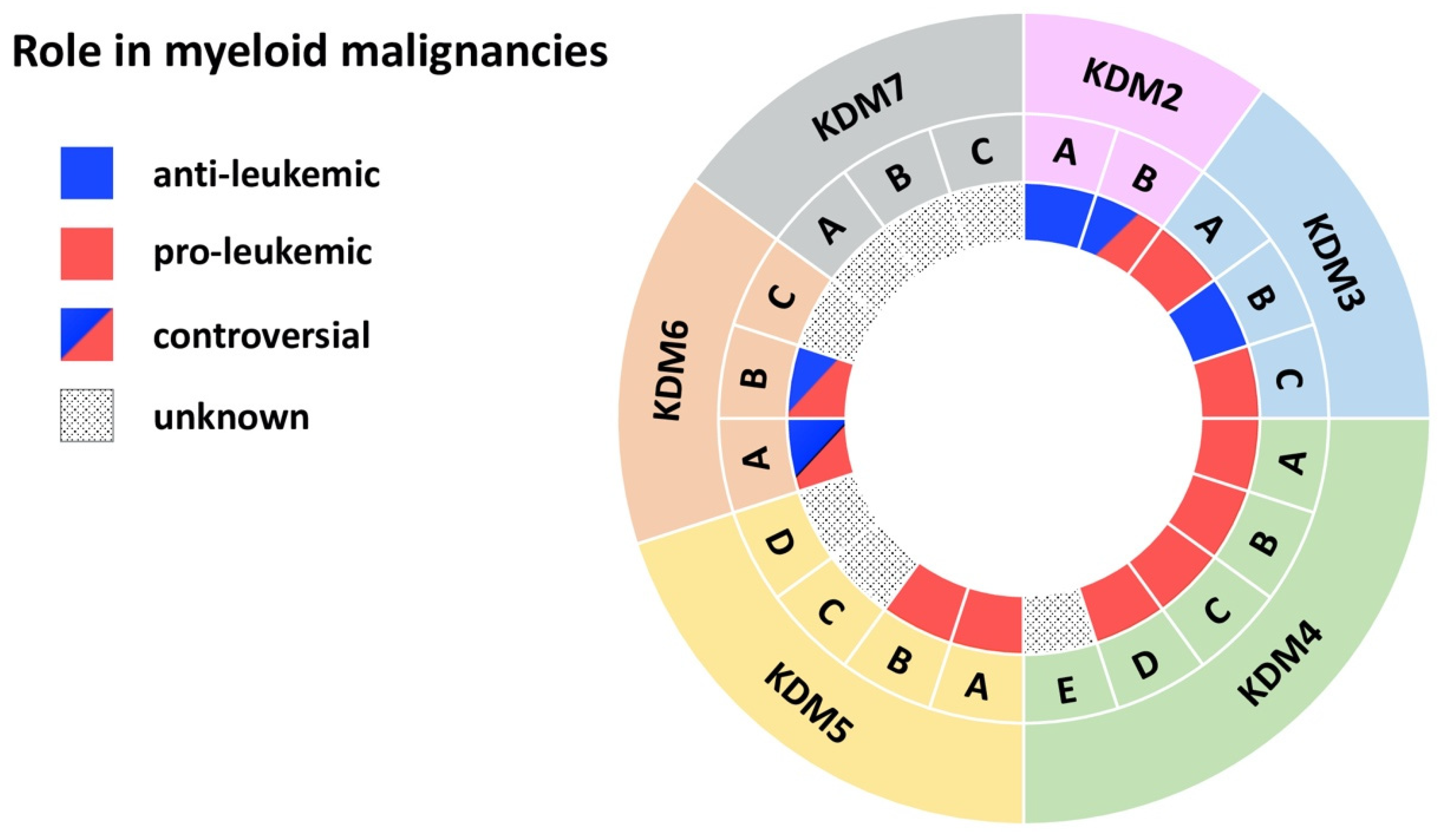

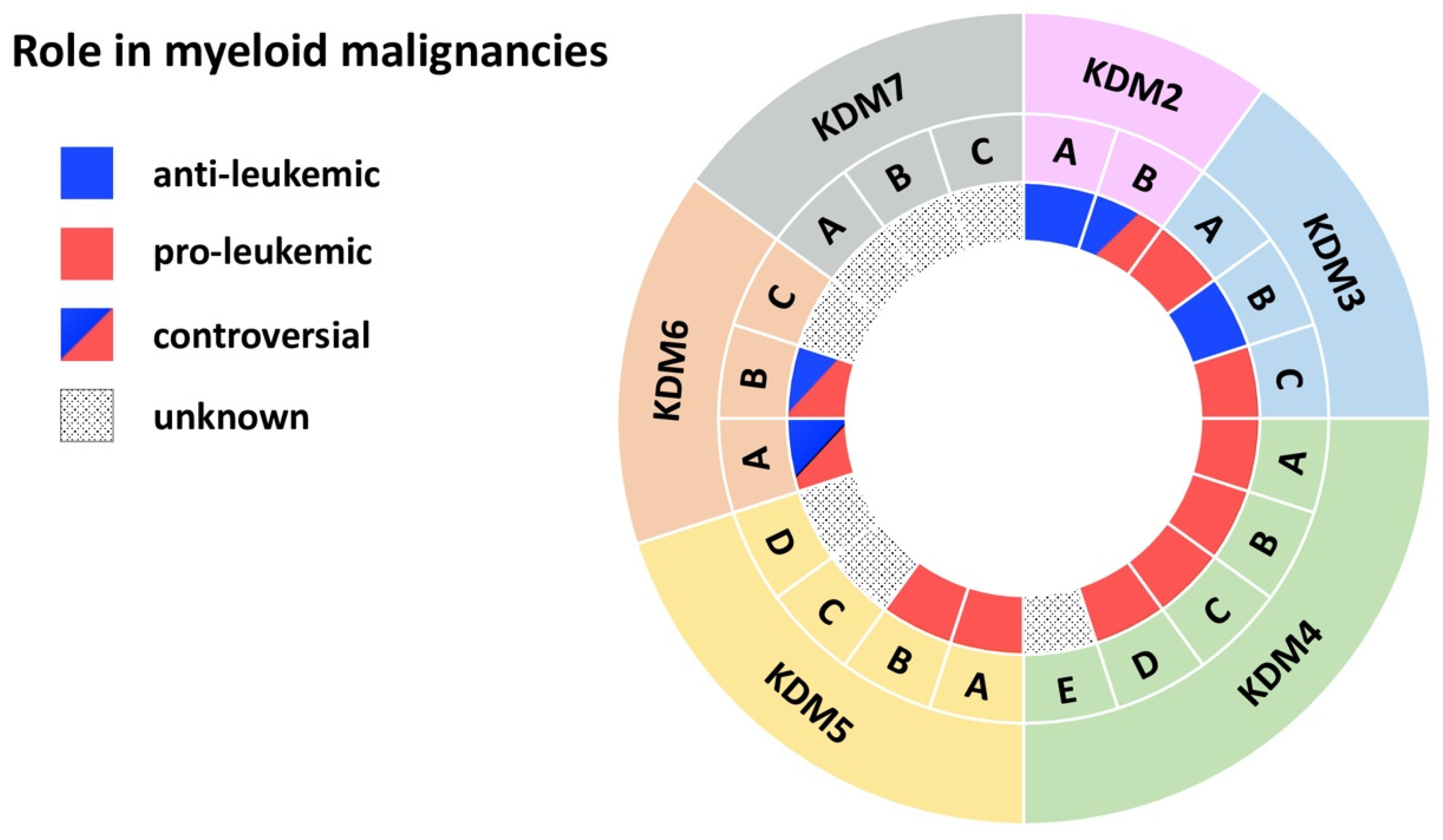
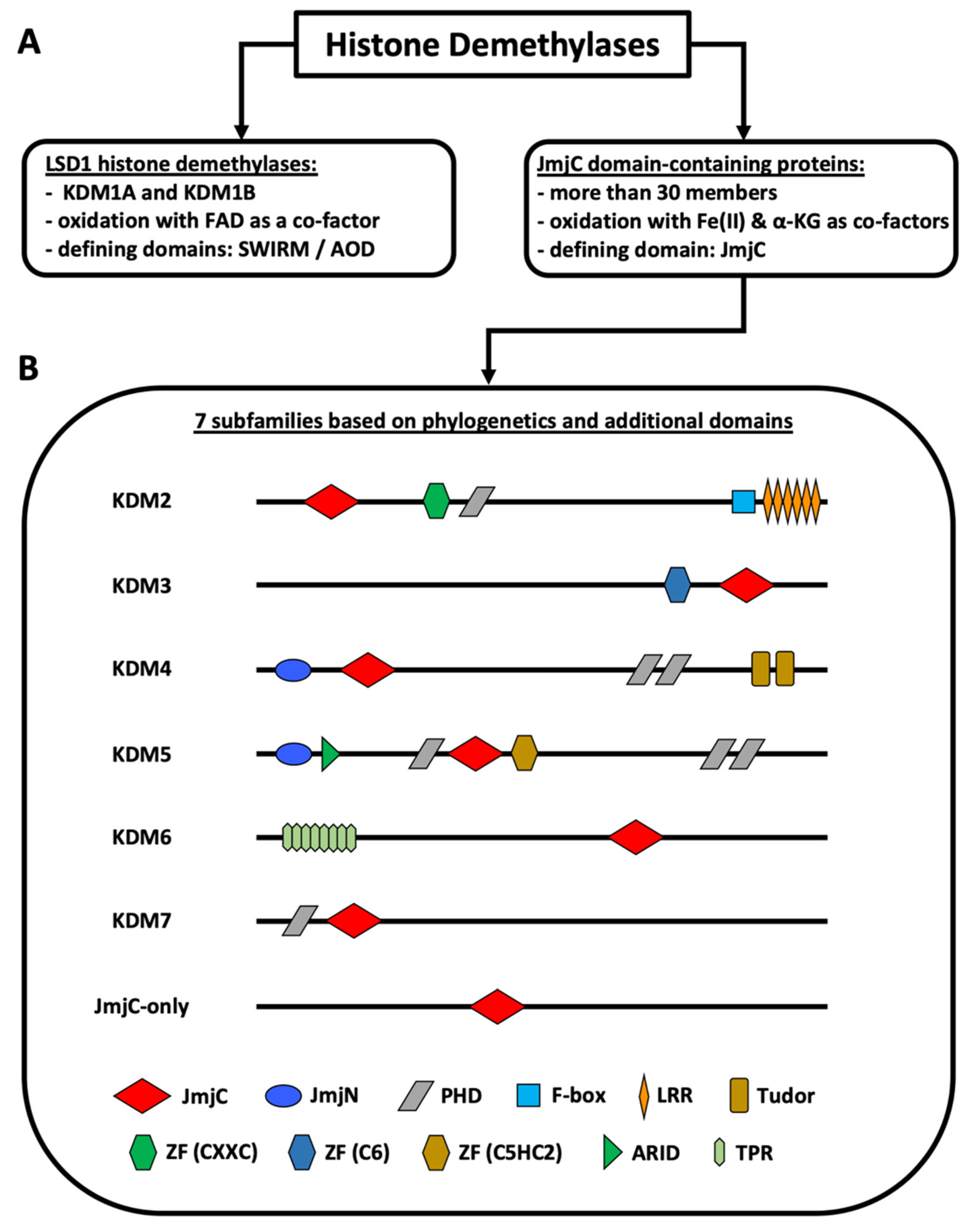{kind=link}
{kind=link}
| Demethylase | Disease | Model | Effect | Mechanism | Reference |
|---|---|---|---|---|---|
| KDM2A | AML | MLL-AF10-induced leukemia in mice | anti-leukemic | KDM2A antagonizes oncogenic LEDGF/ASH1L | [10] |
| AML | Chemically induced leukemia | KDM2A is downregulated in benzene-induced AML cells | [13] | ||
| KDM2B | AML | AML patient cells; MLL-AF9 transduced CD34+ cells; mouse xg models | pro-leukemic | KDM2B as part of the PRC1.1 complex regulates LDHA/PKM independent of H3K27me3 | [16] |
| AML | Tg mouse model with Kdm2b overexpression | KDM2B induces leukemia by increasing expression of Nsg2 and OXPHOS genes | [17] | ||
| AML | AML cell lines; AML CD34+ primary cells | KDM2B promotes cell cycle progression by reducing the tumor suppressor p15 | [11] | ||
| AML | AML patient cells; Hoxa9/Meis1-induced leukemia | KDM2B promotes leukemic transformation by reducing the tumor suppressor p15 | [12] | ||
| MDS | Primary MDS cells, MDS cell lines | anti-leukemic | KDM2B suppresses EZH2 through miRNA let-7b expression | [18] | |
| AML | KrasG12D mice | KDM2B interacts with PRC1/2, increases Irf+Stat, downregulates Hoxa10+Smarca4/Brg1 | [19] | ||
| KDM3A | AML | Primary AML patient cells | pro-leukemic | KDM3A is recruited by Oct1 to the CDX2 promoter to remove repressive H3K9me2 | [20] |
| KDM3B | APL | NB4 APL cell line | anti-leukemic | KDM3B kd enhances proliferation, blocks differentiation, inhibits degradation of PML/RARα | [21] |
| AML | Primary AML patient cells; AML cell lines | KDM3B is downregulated in AML/MDS and overexpression represses colony formation | [22] | ||
| AML | AML cell lines | Expression of KDM2B reduces leukemic growth | [23] | ||
| KDM3C | AML | AML cell lines | pro-leukemic | KDM3C modulators selectively inhibit the growth of leukemic stem cells | [24] |
| Ph+ MPN | K562 and MEG-01 cell lines | KDM3C kd impairs proliferation, viability, and sensitivity towards chemotherapy | [25] | ||
| Ph- MPN | Jak2V617F mice | Loss of Kdm3c is dispensable for disease initiation | [26] | ||
| AML | AML; MLL cell lines | The Kdm3c inhibitor JDI-16 induces apoptosis and differentiation | [27] | ||
| AML | Mouse MLL-AF9 leukemia cells | Loss of Kdm3c activity increases apoptosis+differentiation via RAS/MAPK, JAK-STAT, IL3 | [28] | ||
| AML | HOXA9/MEIS1 bone marrow transplantation model | Kdm3c upregulates key glycolytic and oxidative genes independent of its enzymatic activity | [29] | ||
| Ph-MPN | Primary MPN cells; NFE2 overexpressing mice; MPN cell lines | KDM3C and NFE2 form a positive feedback loop | [30] | ||
| AML | MLL-AF9 and HOXA9 leukemia mice | KDM3C interacts with HOXA9 and supports a HOXA9-controlled gene-expression program | [31] | ||
| AML | AML cell lines | KDM3C is recruited by RUNX1–RUNX1T1 to maintain low H3K9me2 at its target genes | [32] | ||
| AML | MLL-AF9 Tx mouse models; AML cell lines | Depletion of Kdm3c increases apoptosis of leukemic cells | [33] | ||
| KDM4A-C | AML | MLL-AF9 mouse model and cell lines | pro-leukemic | Combined KDM4 demethylase activity promotes survival of leukemic cells and increases expression of Il3ra | [34] |
| KDM4A | AML | Primary AML patient cells; AML cell lines; mouse xg models | pro-leukemic | Loss of KDM4A induces apoptosis and downregulates pro-leukemic gene expression | [35] |
| APL | NB4 APL and other cancer cell lines | KDM4A inhibitors increase H3K9/H3K36 methylation and kill malignant cells | [36] | ||
| KDM4B | AML | MLL-AF9 transduced CD34+ cells; Primary AML patient cells; AML cell lines | pro-leukemic | KDM4B supports proliferation through upregulation of S100A8/9 and loss of KDM4B reduces growth of leukemic cells | [37] |
| KDM4C | AML | Primary AML patient cells; AML cell lines; mouse xg models | pro-leukemic | KDM4C regulates miR-328-3p/CCND2 through MALAT1 resulting in Ara-C resistance | [38] |
| AML | Primary AML patient cells; AML cell lines; mouse xg models | KDM4C upregulates ALKBH5 resulting in increased AXL mRNA stability | [39] | ||
| AML | Leukemic cells with MLL fusions and MOZ-TIF2; mouse xg models | KDM4C regulates target genes of MLL fusions/MOZ-TIF2 via H3K9me3 demethylation | [40] | ||
| AML | AML cell lines | KDM4C mediates oncogenic activity of PRL-3 by reducing H3K9me3 at the Leo1 promoter | [41] | ||
| KDM4D | AML | AML cell lines | pro-leukemic | KDM4D promotes proliferation in AML cells and activates expression of MCL-1 through H3K9me3 demethylation | [42] |
| KDM5A | AML | Mouse NUP98 fusion-induced leukemia | pro-leukemic | NUP98-KDM5A (PHD finger) fusions induces differentiation arrest and leukemia | [43] |
| CML | K562 cells | KDM5A kd in CML-BP stimulates leukemia cell differentiation and inhibits cell proliferation | [44] | ||
| CML | K562 cells, primary patient samples | miR-181d downregulates KDM5A which inhibits NF-κB subunit, p65 | [45] | ||
| KDM5B | AML/CML | CD34+ cells, AML and CML cell lines | pro-leukemic | KDM5B is highly expressed AML/CML cells, kd reduced leukemia colony-forming abilities | [46] |
| AML | Mouse MLL-AF9/10 leukemia cells, MLLr patient samples | KDM5B negatively regulates leukemogenesis | [47] | ||
| AML | Clinical data | KDM5B expression predict survival | [48] | ||
| AML | Mouse | KDM5B is required for hematopoietic stem cell self-renewal | [49,50] | ||
| KDM5C | AML | Primary AML patient cells (M5) | unknown | KDM5C is overexpressed in pediatric AML (M5) | [51] |
| AML | Primary AML patient cells | KDM5C is mutated and enriched in chemotherapy-resistant pediatric leukemia | [52] | ||
| KDM6A | CMML | KDM6A ko mice | anti-leukemic | Loss of KDM6A causes an CMML-like disease | [53] |
| MDS | KDM6A ko mice | Loss of KDM6A causes myelodysplasia | [54] | ||
| AML | KDM6A ko mice, AML cell lines | KDM6A ko causes COMPASS complex malfunctioning with upregulation of ETS signaling | [55] | ||
| AML | Primary AML patient cells; AML cell lines | Loss of KDM6A confers cytarabine resistance through ENT1 downregulation | [56] | ||
| CML | Primary CML patient cells; CML cell lines | pro-leukemic | KDM6A promotes imatinib-resistance through upregulation of TRKA | [57] | |
| AML/CML | AML and CML cell lines | KDM6A depletion reduces Runx1, Mll1 and Scl expression and impairs proliferation | [58] | ||
| AML | Primary AML patient cells; AML cell lines | KDM6A promotes cancer cell survival via upregulation of DOCK5/8 | [59] | ||
| KDM6B | MDS | Primary MDS patient CD34+ cells | pro-leukemic | Inhibition of KDM6B resulted in an increase in erythroid colonies in MDS | [60] |
| AML | Clinical data | KDM6B is overexpressed in AML and correlates with a. poor survival | [61] | ||
| MDS/CMML | Tg KDM6B overexpression in mice | KDM6B overexpression showed features of MDS in mice | [62] | ||
| AML | Kdm6b ko (VAVCre, MxCre, ERT2-Cre) | Loss of Kdm6b reduced HSCs and attenuates MLL-AF9-induced AML | [63] | ||
| AML | AML cell lines | KDM6B kd reduced the proliferation and increased chemo-sensitivity | [64] | ||
| AML | HL-60; primary patient samples; PML-RARα-, AML1-ETO9a, or MLL-AF9 tg mice | anti-leukemic | KDM6B exerts anti-AML effect by directly modulating H3K4 and H3K27 | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staehle, H.F.; Pahl, H.L.; Jutzi, J.S. The Cross Marks the Spot: The Emerging Role of JmjC Domain-Containing Proteins in Myeloid Malignancies. Biomolecules 2021, 11, 1911. https://doi.org/10.3390/biom11121911
Staehle HF, Pahl HL, Jutzi JS. The Cross Marks the Spot: The Emerging Role of JmjC Domain-Containing Proteins in Myeloid Malignancies. Biomolecules. 2021; 11(12):1911. https://doi.org/10.3390/biom11121911
Chicago/Turabian StyleStaehle, Hans Felix, Heike Luise Pahl, and Jonas Samuel Jutzi. 2021. "The Cross Marks the Spot: The Emerging Role of JmjC Domain-Containing Proteins in Myeloid Malignancies" Biomolecules 11, no. 12: 1911. https://doi.org/10.3390/biom11121911
APA StyleStaehle, H. F., Pahl, H. L., & Jutzi, J. S. (2021). The Cross Marks the Spot: The Emerging Role of JmjC Domain-Containing Proteins in Myeloid Malignancies. Biomolecules, 11(12), 1911. https://doi.org/10.3390/biom11121911