
Protective Effect of Osmundacetone against Neurological Cell Death Caused by Oxidative Glutamate Toxicity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Isolation of Compound
2.3. Cell Culture
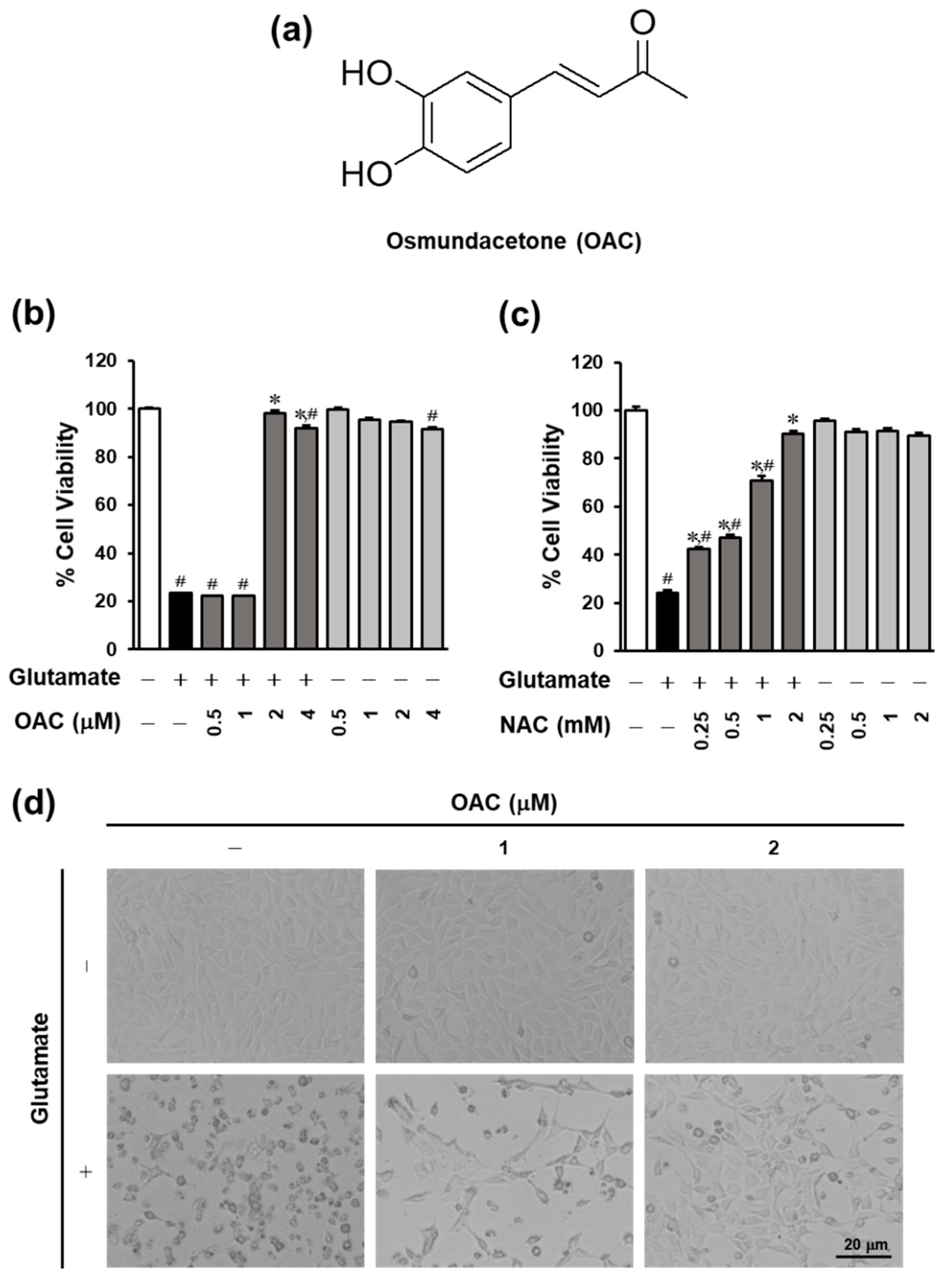
2.4. Cell Viability Assay
2.5. DPPH Radical Scavenging Assay
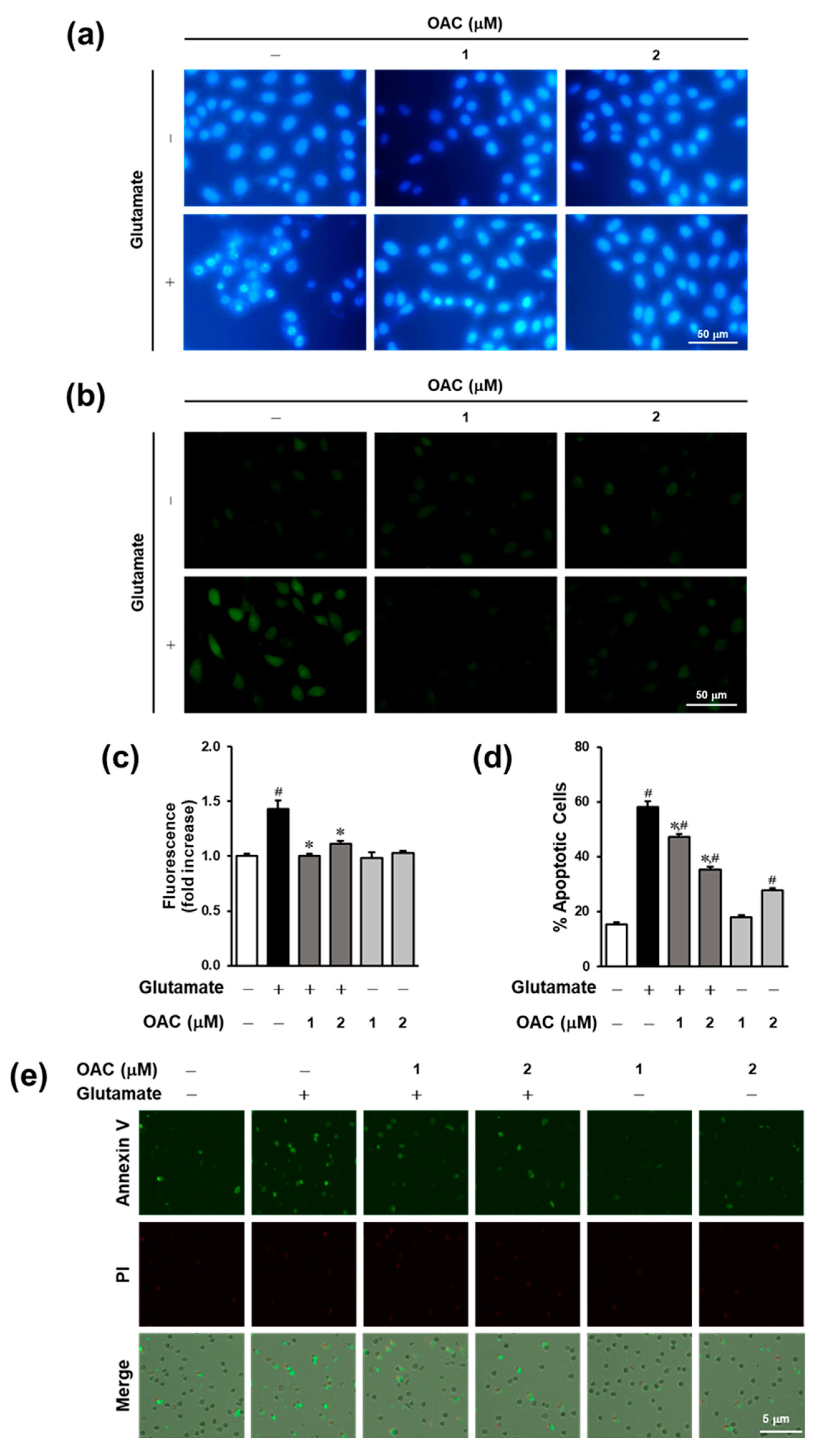
2.6. ROS Assay
2.7. Ca2+ Staining
2.8. Hoechst 33342 Staining
2.9. Image-Based Apoptosis Detection Assay
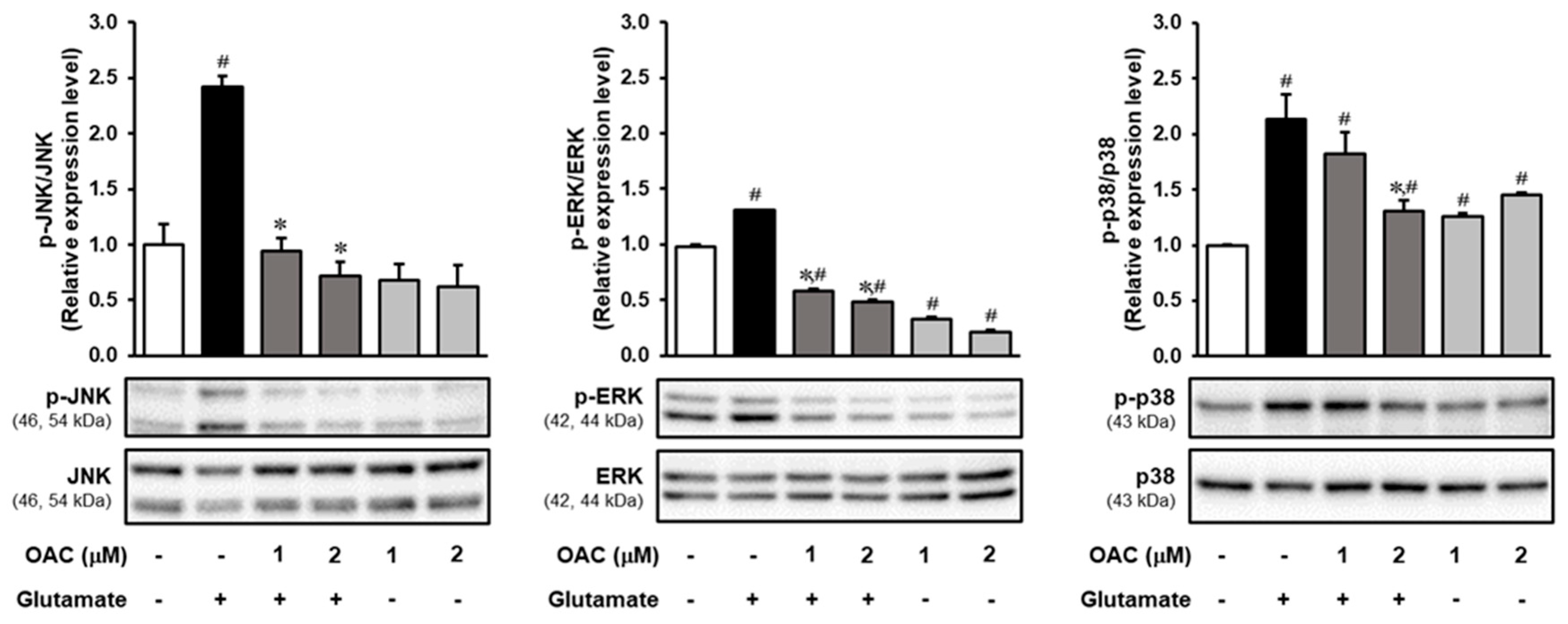
2.10. Western Blotting Analysis
2.11. Statistical Analysis
3. Results
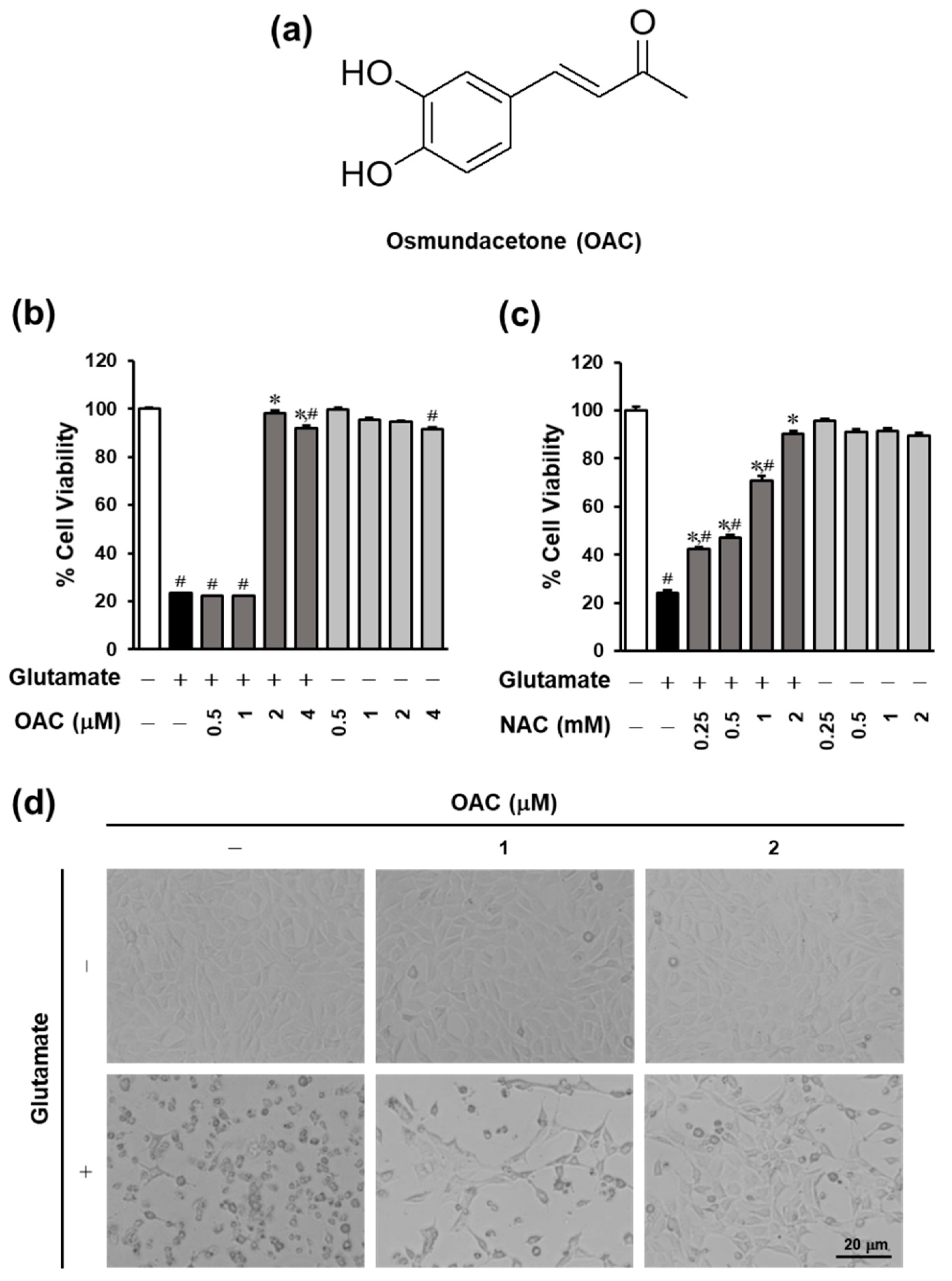
3.1. Neuroprotective Effect of OAC on HT22 Cells
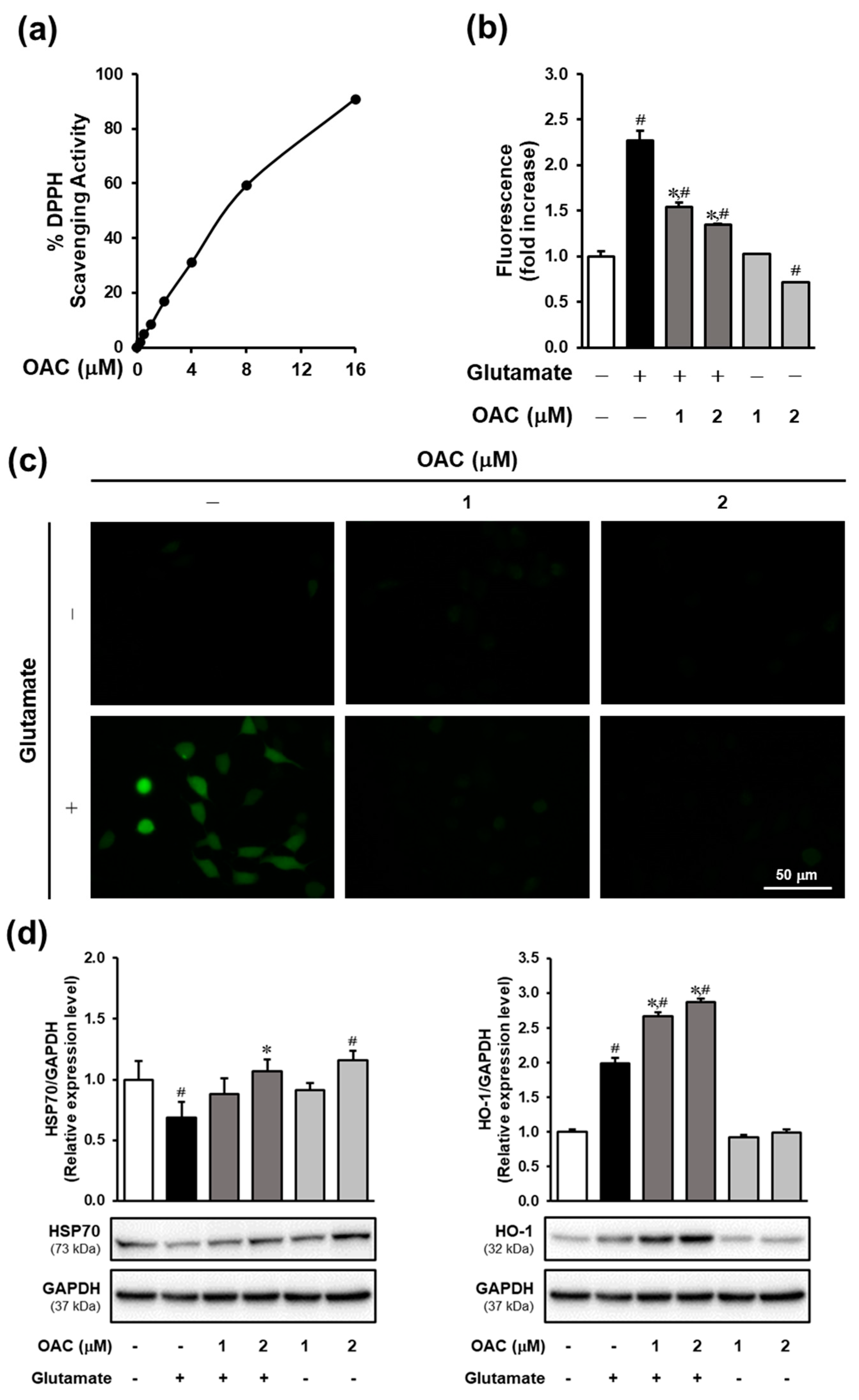
3.2. Effect of OAC Against Oxidative Stress
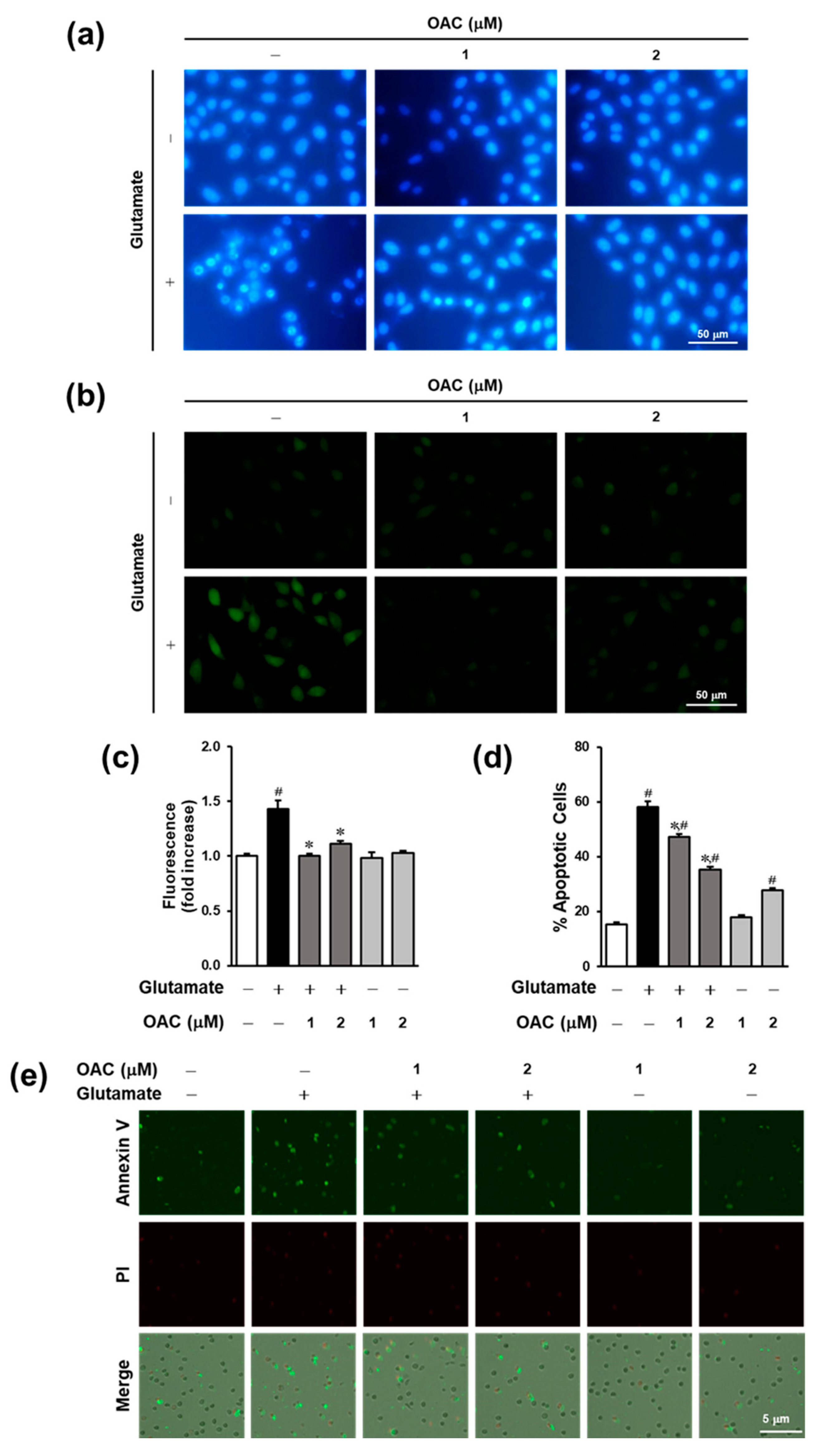
3.3. Anti-Apoptotic Effect of OAC on HT22 Cells
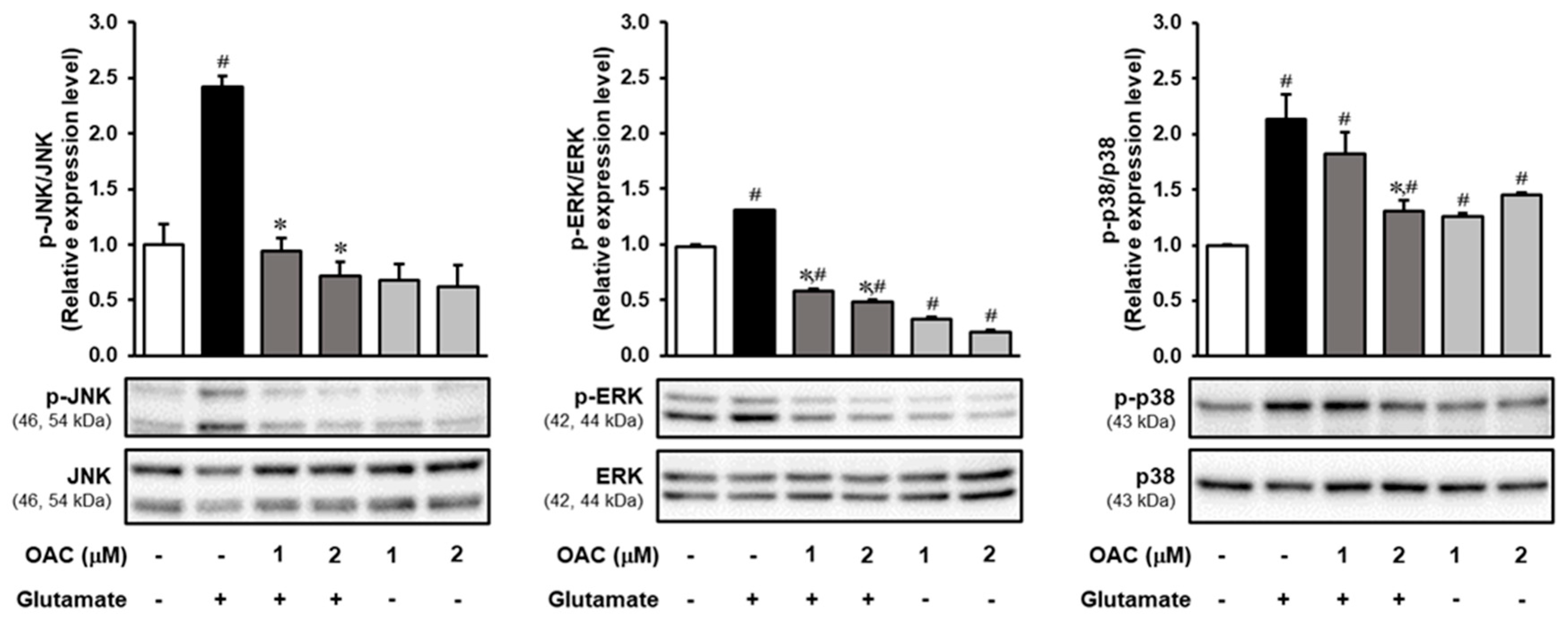
3.4. Regulatory Effect of OAC on the Phosphorylation of MAPKs Induced by Glutamate
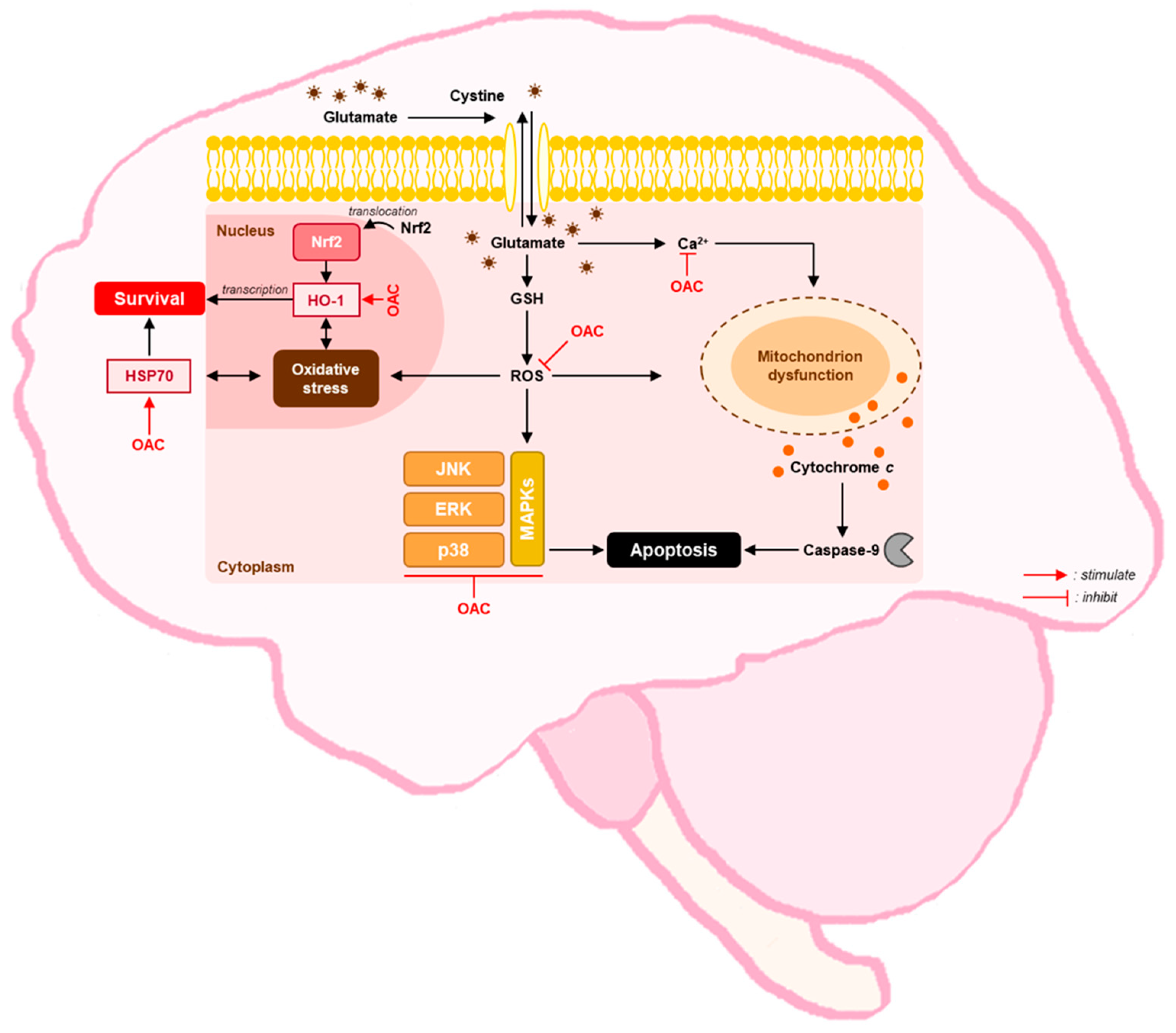
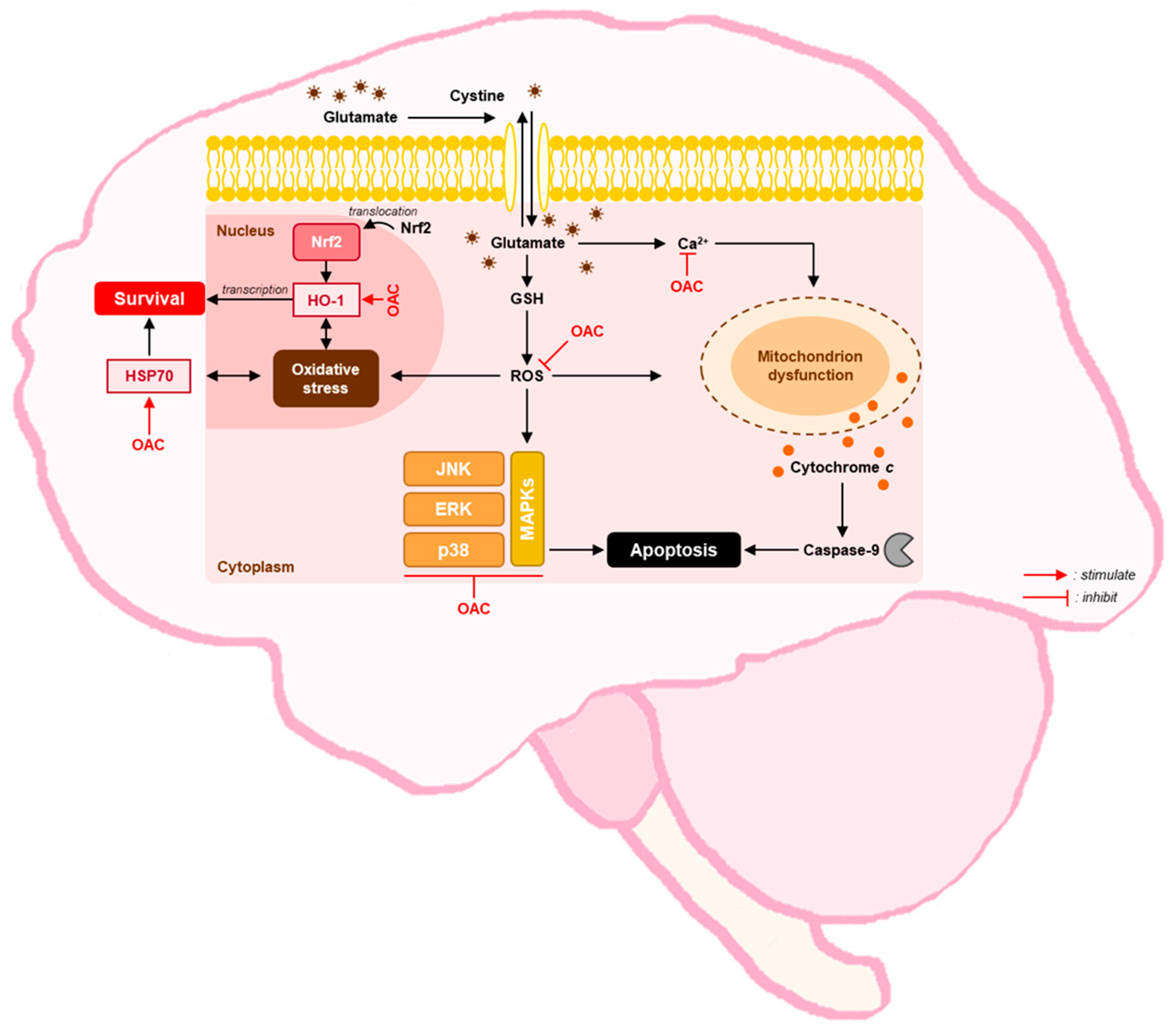
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO. Neurological Disorders: Public Health Challenges; WHO: Geneva, Switzerland, 2006; pp. 35–37. [Google Scholar]
- Beghi, E.; Giussani, G.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Adib, M.G.; Agrawal, S.; Alahdab, F.; et al. Global, regional, and national burden of epilepsy, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 357–375. [Google Scholar] [CrossRef] [Green Version]
- WHO. Global Action Plan on the Public Health Response to Dementia 2017–2025; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A Double-Blind, Placebo-Controlled Randomized Clinical Trial of Alpha-Tocopherol (Vitamin E) in the Treatment of Amyotrophic Lateral Sclerosis. ALS Riluzole-Tocopherol Study Group. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2001, 2, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Arlt, S.; Muller-Thomsen, T.; Beisiegel, U.; Kontush, A. Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem. Res. 2012, 37, 2706–2714. [Google Scholar] [CrossRef]
- Gubandru, M.; Margina, D.; Tsitsimpikou, C.; Goutzourelas, N.; Tsarouhas, K.; Ilie, M.; Tsatsakis, A.M.; Kouretas, D. Alzheimer’s disease treated patients showed different patterns for oxidative stress and inflammation markers. Food Chem. Toxicol. 2013, 61, 209–214. [Google Scholar] [CrossRef]
- Snalina, N.; Alessenko, A.; Gavrilova, S.; Gurianova, S.; Prochorov, A.; Kononova, E.; Fedorova, Y. Memantine changes lipids spectrum and lipid peroxidation in animal brain and plasma of patients with Alzheimer’s disease. FEBS J. 2014, 281, 65–784. [Google Scholar] [CrossRef] [Green Version]
- Fonnum, F. Glutamate: A neurotransmitter in mammalian brain. J. Neurochem. 1984, 42, 1–11. [Google Scholar] [CrossRef]
- Tapiero, H.; Mathe, G.; Couvreur, P.; Tew, K.D., II. Glutamine and glutamate. Biomed. Pharmacother. 2002, 56, 446–457. [Google Scholar] [CrossRef]
- Herrera, F.; Sainz, R.M.; Mayo, J.C.; Martín, V.; Antolín, I.; Rodriguez, C. Glutamate induces oxidative stress not mediated by glutamate receptors or cystine transporters: Protective effect of melatonin and other antioxidants. J. Pineal. Res. 2001, 31, 356–362. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L.; Snyder, S.H. A novel neuronal messenger molecule in brain: The free radical, nitric oxide. Ann. Neurol. 1992, 32, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Dumuis, A.; Sebben, M.; Haynes, L.; Pin, J.P.; Bockaert, J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature 1988, 336, 68–70. [Google Scholar] [CrossRef]
- Bannai, S.; Kitamura, E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 1980, 255, 2372–2376. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravindran, P.N. The Encyclopedia of Herbs and Spices; CABI: Wallingford, UK, 2017. [Google Scholar]
- Yip, L.; Hudson, J.B.; Towers, G.N. Towers GH. Isolation of the anthropogenic compound fluoranthene in a screening of Chinese medicinal plants for antiviral compounds. Planta Med. 1995, 61, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, J.; Yang, L.; Yang, F.; Ge, H.; Zhao, C.; Zhang, L.; Zu, Y. Evaluation of antioxidant activities of aqueous extracts and fractionation of different parts of Elsholtzia ciliata. Molecules 2012, 17, 5430–5441. [Google Scholar] [CrossRef]
- Pudziuvelyte, L.; Liaudanskas, M.; Jekabsone, A.; Sadauskiene, I.; Bernatoniene, J. Elsholtzia ciliata (Thunb.) Hyl. Extracts from Different Plant Parts: Phenolic Composition, Antioxidant, and Anti-Inflammatory Activities. Molecules 2020, 25, 1153. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.H.; Yoo, J.S.; Lee, H.S.; Kwon, T.K.; Shin, T.Y.; Kim, S.H. Elsholtzia ciliata inhibits mast cell-mediated allergic inflammation: Role of calcium, p38 mitogen-activated protein kinase and nuclear factor-{kappa}B. Exp. Biol. Med. (Maywood) 2011, 236, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kim, Y.J.; Seo, C.S.; Kim, H.T.; Park, S.R.; Lee, M.Y.; Jung, J.Y. Elsholtzia ciliata (Thunb.) Hylander attenuates renal inflammation and interstitial fibrosis via regulation of TGF-ss and Smad3 expression on unilateral ureteral obstruction rat model. Phytomedicine 2016, 23, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Le, T.B.; Beaufay, C.; Nghiem, D.T.; Mingeot-Leclercq, M.P.; Quetin-Leclercq, J. In Vitro Anti-Leishmanial Activity of Essential Oils Extracted from Vietnamese Plants. Molecules 2017, 22, 1071. [Google Scholar] [CrossRef] [Green Version]
- Nugroho, A.; Park, J.H.; Choi, J.S.; Park, K.S.; Hong, J.P.; Park, H.J. Structure determination and quantification of a new flavone glycoside with anti-acetylcholinesterase activity from the herbs of Elsholtzia ciliata. Nat. Prod. Res. 2019, 33, 814–821. [Google Scholar] [CrossRef]
- Choi, M.S.; Choi, B.S.; Kim, S.H.; Pak, S.C.; Jang, C.H.; Chin, Y.W.; Kim, Y.M.; Kim, D.I.; Jeon, S.; Koo, B.S. Essential Oils from the Medicinal Herbs Upregulate Dopamine Transporter in Rat Pheochromocytoma Cells. J. Med. Food 2015, 18, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Hwang, B.S.; Lee, I.K.; Seo, G.S.; Yun, B.S. Chemical Constituents of the Culture Broth of Phellinus linteus and Their Antioxidant Activity. Mycobiology 2015, 43, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.S.; Lee, M.S.; Lee, S.W.; Lee, I.K.; Seo, G.S.; Choi, H.J.; Yun, B.S. Neuraminidase Inhibitors from the Fermentation Broth of Phellinus linteus. Mycobiology 2014, 42, 189–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, W.; Deng, J.S.; Huang, S.S.; Li, P.Y.; Liang, Y.C.; Huang, G.J. 3, 4-dihydroxybenzalacetone attenuates lipopolysaccharide-induced inflammation in acute lung injury via down-regulation of MMP-2 and MMP-9 activities through suppressing ROS-mediated MAPK and PI3K/AKT signaling pathways. Int. Immunopharmacol. 2017, 50, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwon, H.J.; Kim, B.W. Protective Effect of 4-(3,4-Dihydroxyphenyl)-3-Buten-2-One from Phellinus linteus on Naproxen-Induced Gastric Antral Ulcers in Rats. J. Microbiol. Biotechnol. 2016, 26, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Gunjima, K.; Tomiyama, R.; Takakura, K.; Yamada, T.; Hashida, K.; Nakamura, Y.; Konishi, T.; Matsugo, S.; Hori, O. 3,4-dihydroxybenzalacetone protects against Parkinson’s disease-related neurotoxin 6-OHDA through Akt/Nrf2/glutathione pathway. J. Cell. Biochem. 2014, 115, 151–160. [Google Scholar] [CrossRef]
- Tomiyama, R.; Takakura, K.; Takatou, S.; Le, T.M.; Nishiuchi, T.; Nakamura, Y.; Konishi, T.; Matsugo, S.; Hori, O. 3,4-dihydroxybenzalacetone and caffeic acid phenethyl ester induce preconditioning ER stress and autophagy in SH-SY5Y cells. J. Cell. Physiol. 2018, 233, 1671–1684. [Google Scholar] [CrossRef]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bhavnani, B.R. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006, 7, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, M.; Ito, Y. N-acetylcysteine and neurodegenerative diseases: Basic and clinical pharmacology. Cerebellum 2007, 6, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.W.M.; Maher, P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. 2002, 71, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Rona, G.; Giffard, M.A.Y. Many mechanisms for Hsp70 protection from cerebral ischemia. J. Neurosurg. Anesthesiol. 2004, 16, 53–61. [Google Scholar]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gary, L.; Johnson, R.L. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar]
- Li, J.O.W.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh, T.A.; Seo, Y.H.; Choi, S.; Lee, J.; Kang, K.S. Protective Effect of Osmundacetone against Neurological Cell Death Caused by Oxidative Glutamate Toxicity. Biomolecules 2021, 11, 328. https://doi.org/10.3390/biom11020328
Trinh TA, Seo YH, Choi S, Lee J, Kang KS. Protective Effect of Osmundacetone against Neurological Cell Death Caused by Oxidative Glutamate Toxicity. Biomolecules. 2021; 11(2):328. https://doi.org/10.3390/biom11020328
Chicago/Turabian StyleTrinh, Tuy An, Young Hye Seo, Sungyoul Choi, Jun Lee, and Ki Sung Kang. 2021. "Protective Effect of Osmundacetone against Neurological Cell Death Caused by Oxidative Glutamate Toxicity" Biomolecules 11, no. 2: 328. https://doi.org/10.3390/biom11020328
APA StyleTrinh, T. A., Seo, Y. H., Choi, S., Lee, J., & Kang, K. S. (2021). Protective Effect of Osmundacetone against Neurological Cell Death Caused by Oxidative Glutamate Toxicity. Biomolecules, 11(2), 328. https://doi.org/10.3390/biom11020328