
Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies

Abstract
:1. Introduction
2. Material and Methods
2.1. Identification of Driver Regions in Cancer-Associated Proteins
2.2. Structural Categorization of Driver Regions
2.3. System-Level Analyses
3. Results
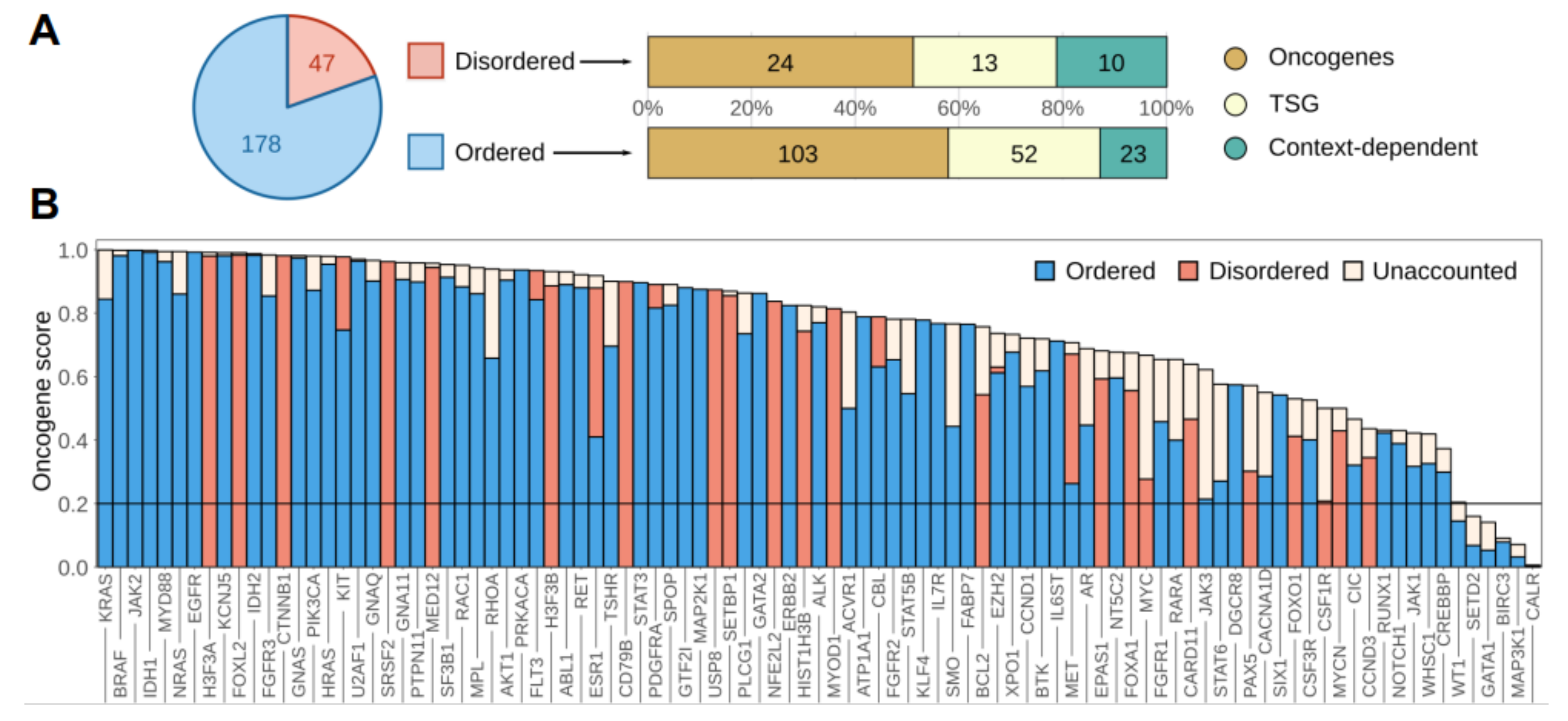
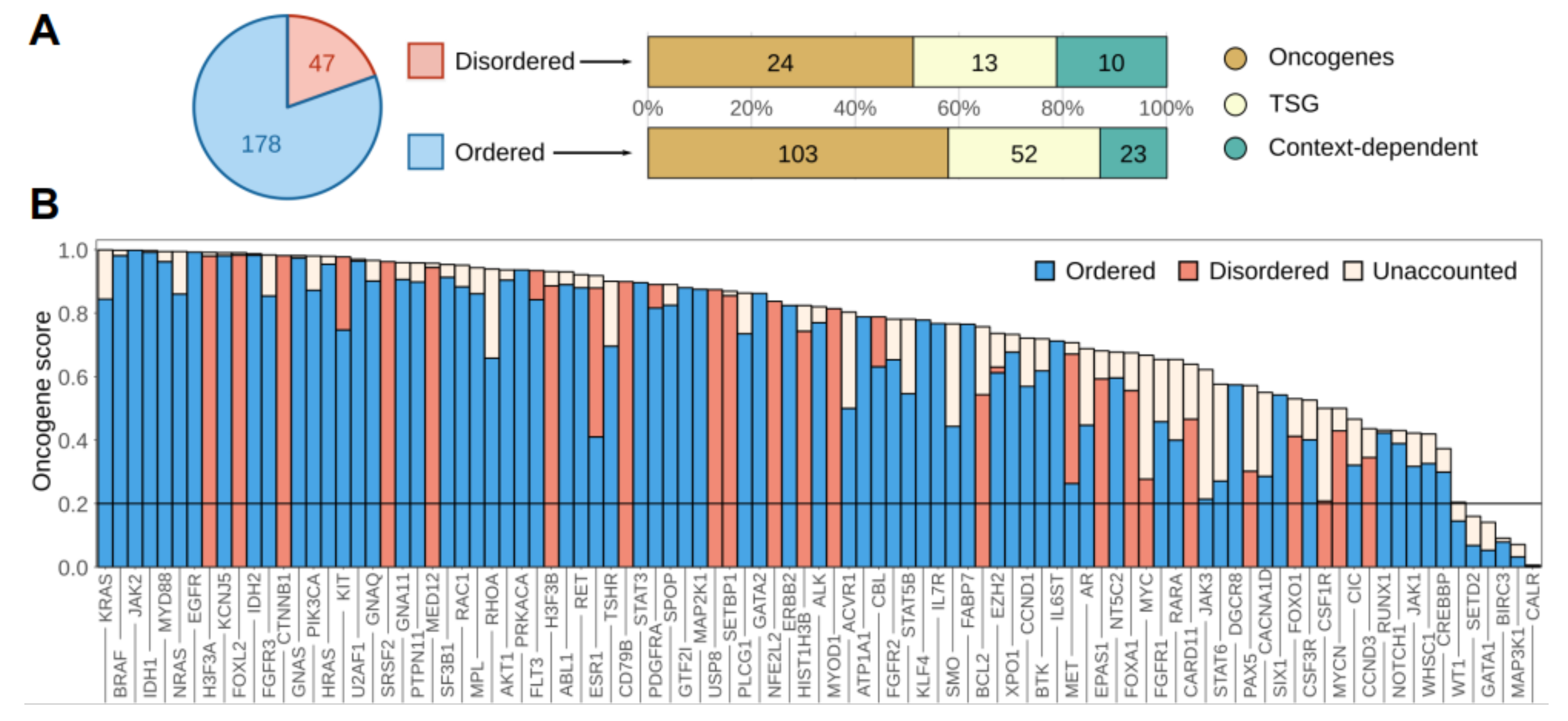
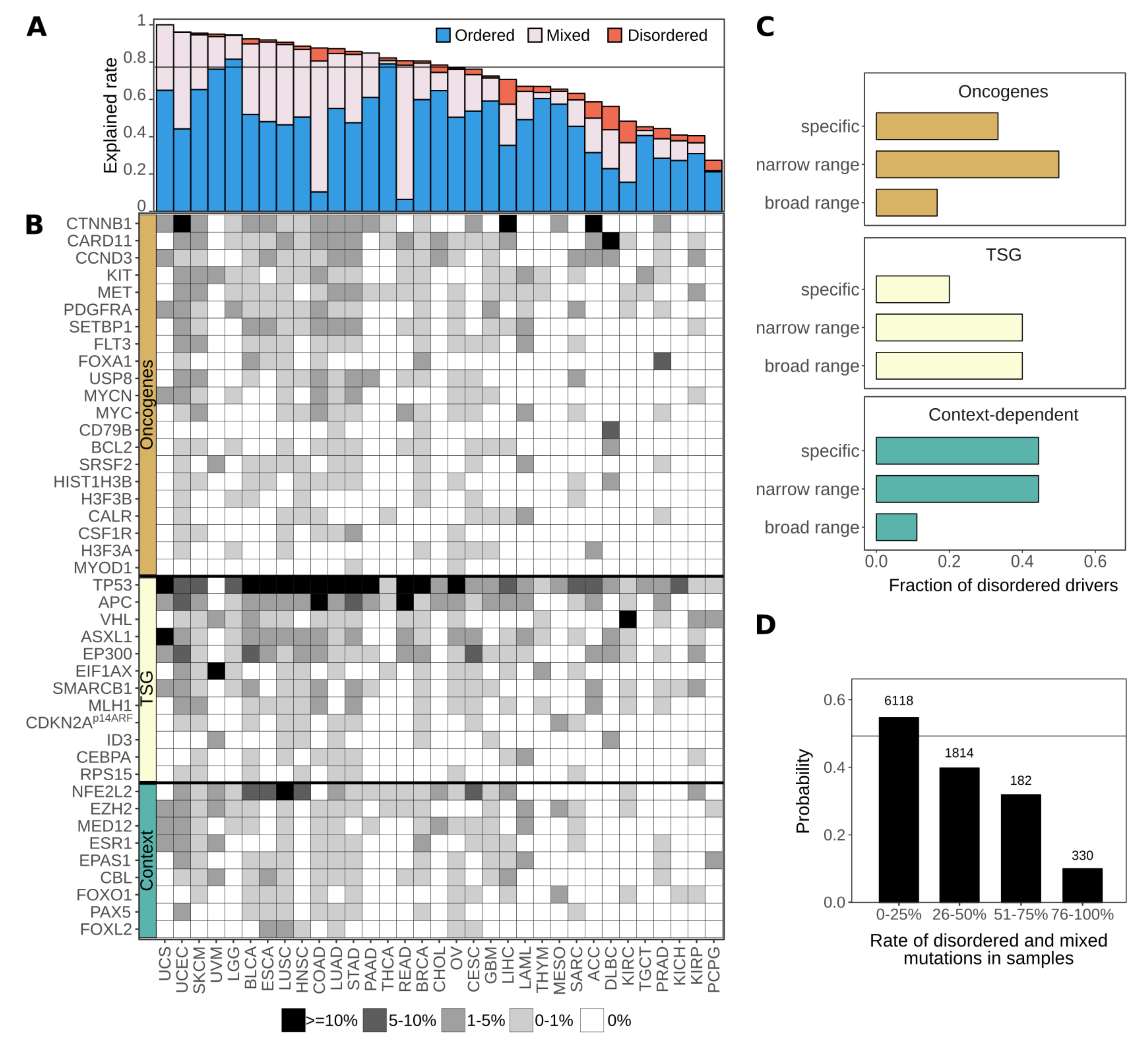
3.1. Disordered Protein Modules Are Targets for Tumorigenic Mutations
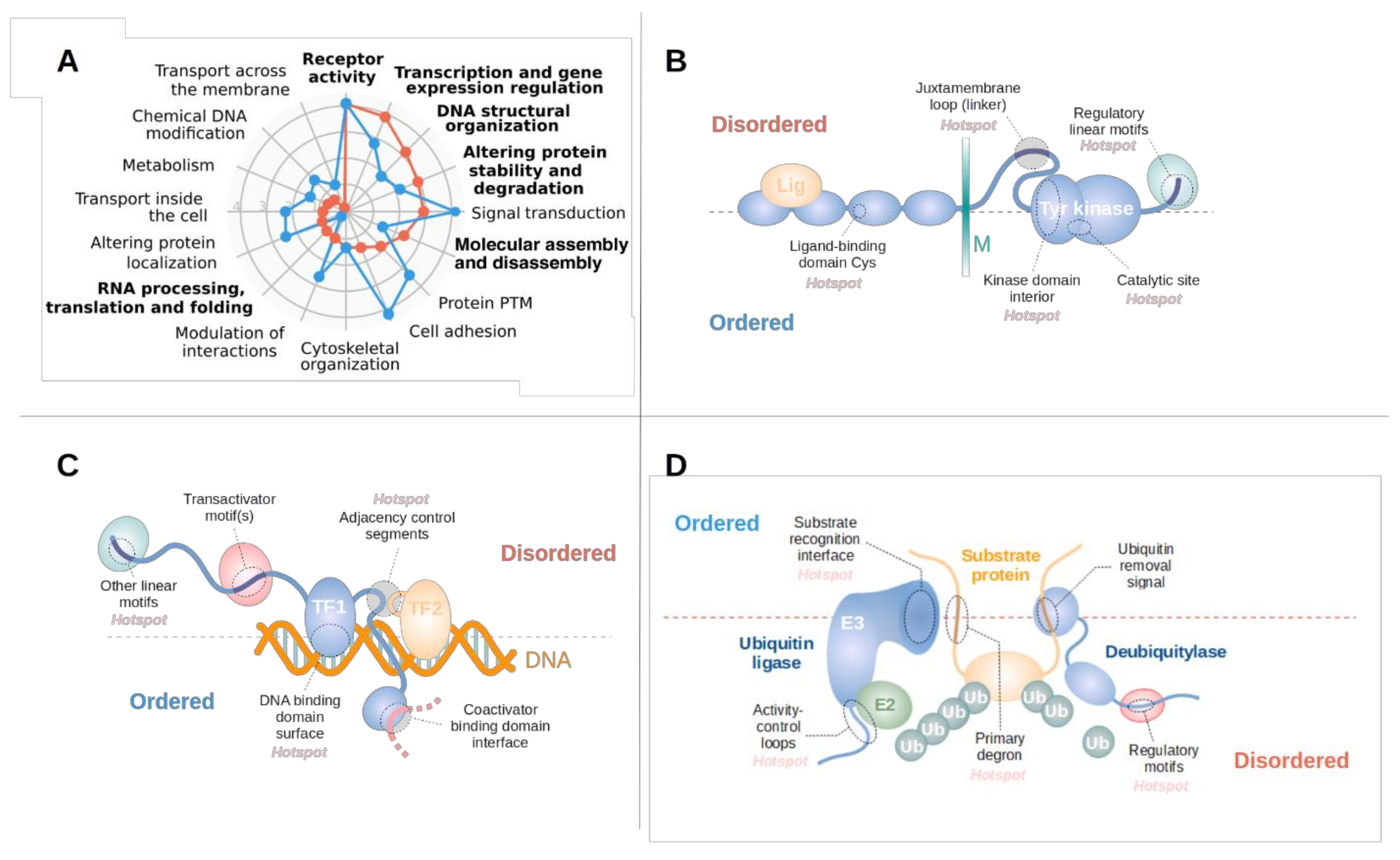
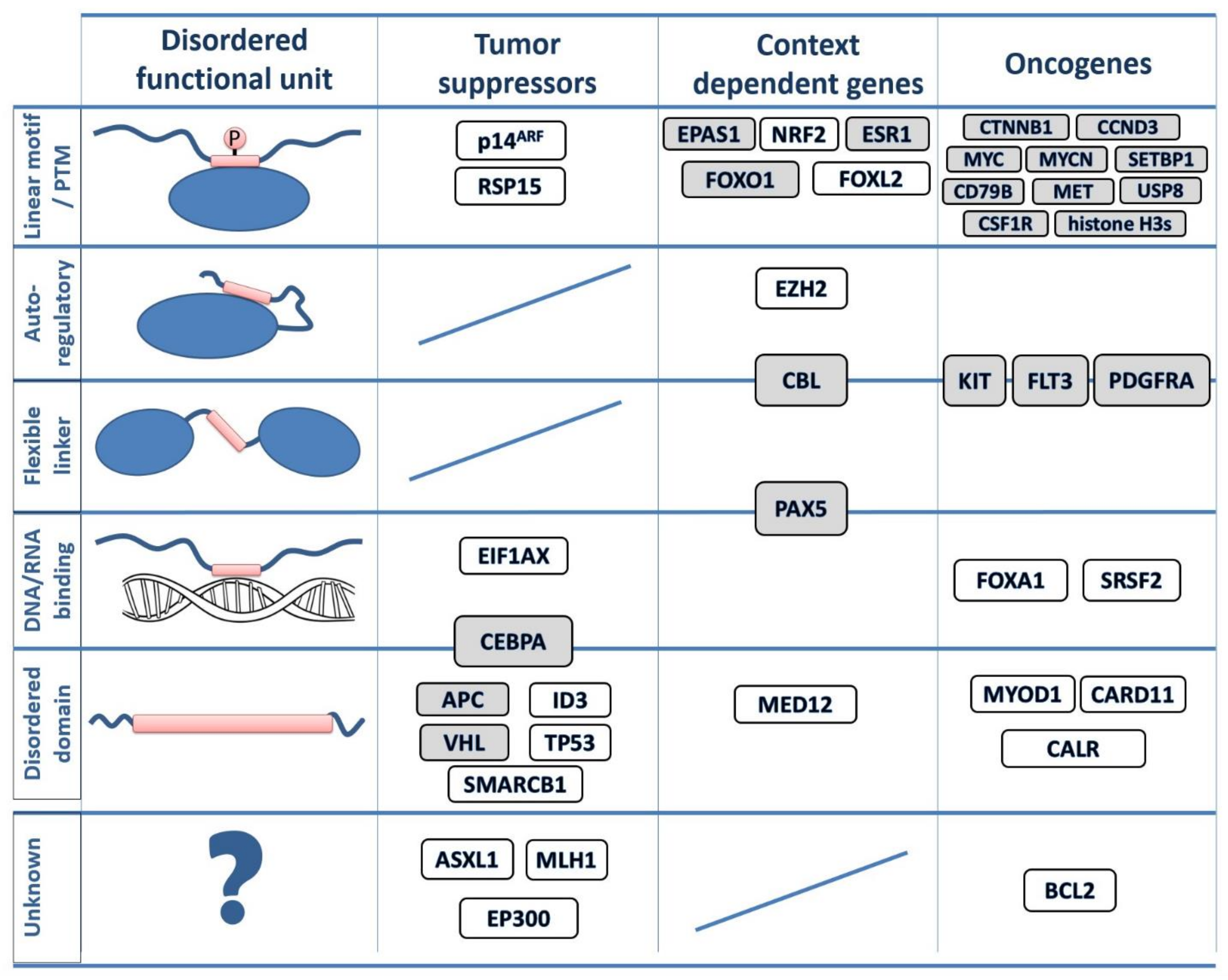
3.2. Disordered Drivers Function via Distinct Molecular Mechanisms
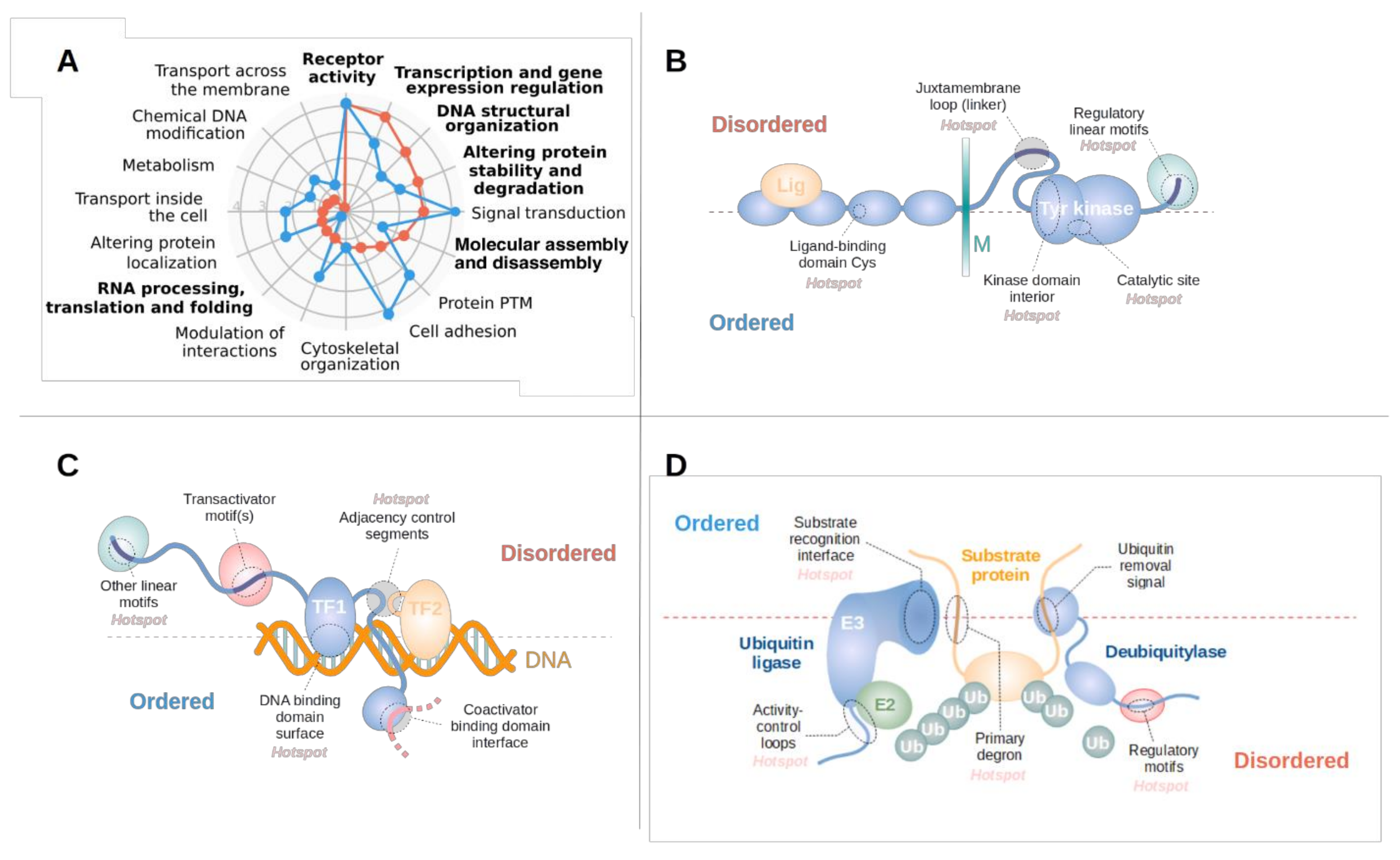
3.3. Disordered Driver Mutations Preferentially Modulate Receptor Tyrosine Kinases, DNA-Processing and The Degradation Machinery
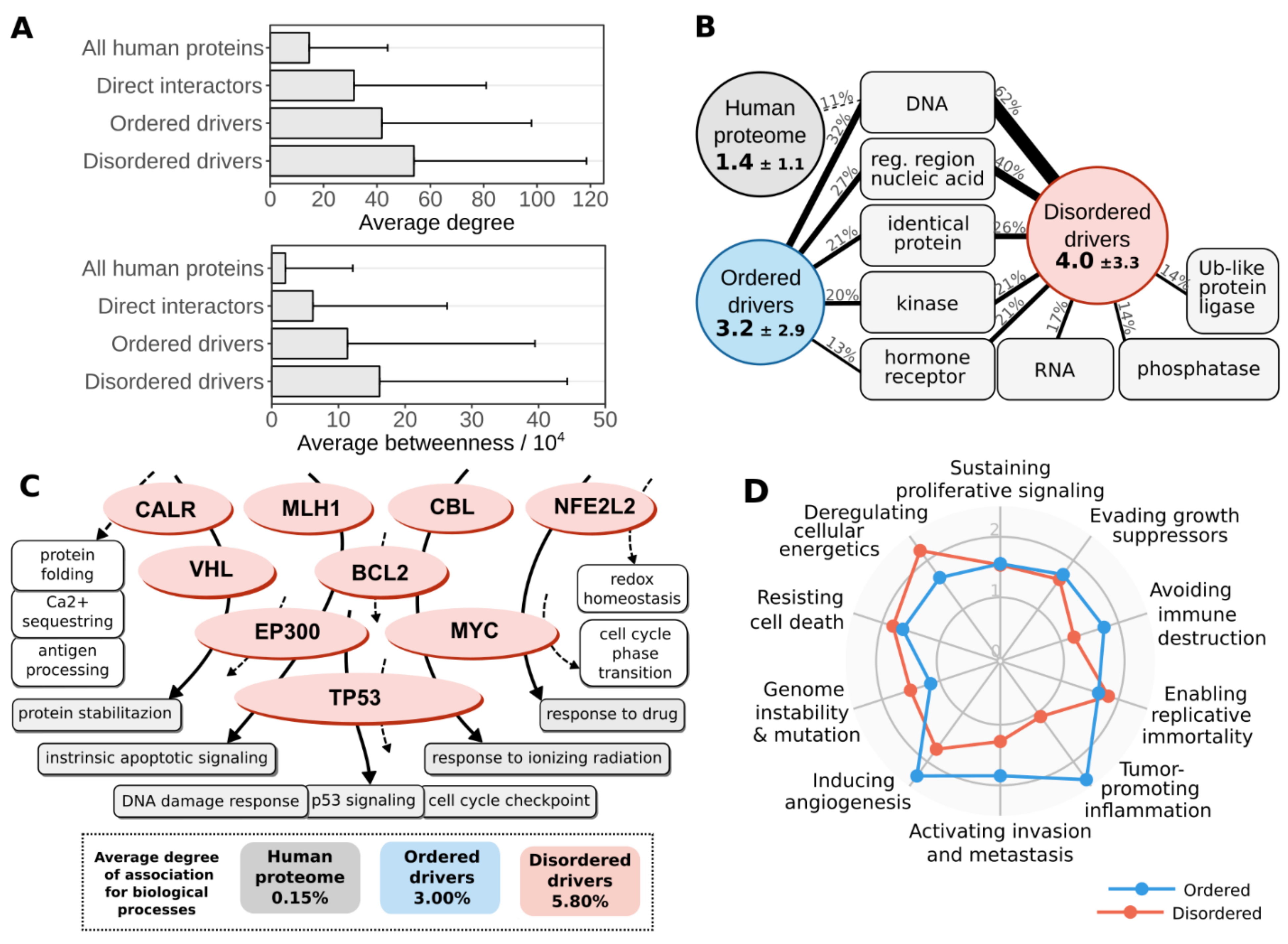
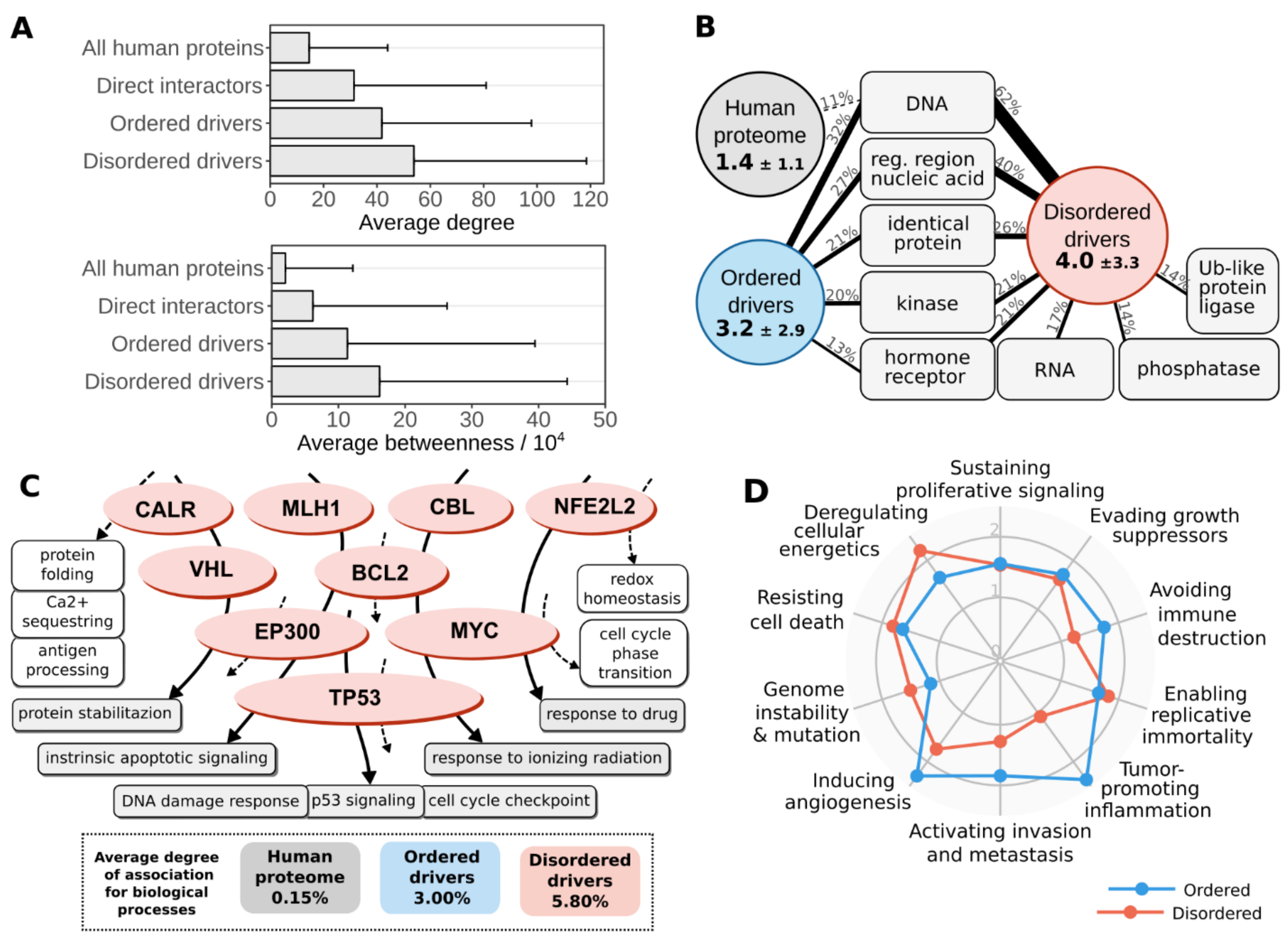
3.4. Disordered Mutations Give Rise to Cancer Hallmarks by Targeting Central Elements of Biological Networks
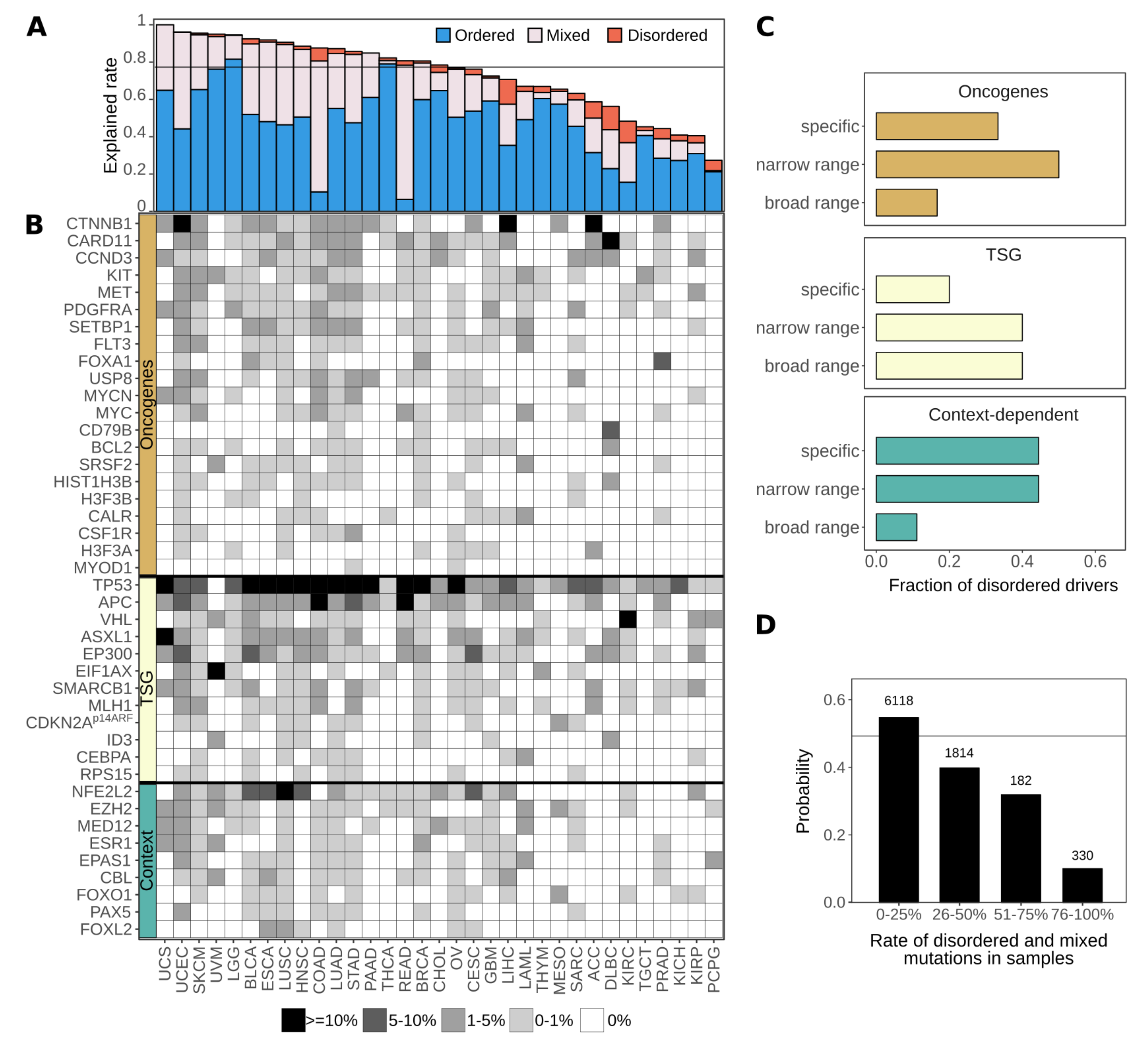
3.5. Disordered Drivers Can Be the Dominant Players at The Patient Sample Level
3.6. Cancer Incidences Arising through Disordered Drivers Lack Effective Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nussinov, R.; Jang, H.; Tsai, C.-J.; Cheng, F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019, 15, e1006658. [Google Scholar]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [Green Version]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short Linear Motifs: Ubiquitous and Functionally Diverse Protein Interaction Modules Directing Cell Regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.W.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic Disorder in Cell-signaling and Cancer-associated Proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Pajkos, M.; Mészáros, B.; Simon, I.; Dosztányi, Z. Is there a biological cost of protein disorder? Analysis of cancer-associated mutations. Mol. BioSyst. 2011, 8, 296–307. [Google Scholar] [CrossRef]
- Hegyi, H.; Buday, L.; Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Comput. Biol. 2009, 5, e1000552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavouri, T.; Semple, J.I.; Garcia-Verdugo, R.; Lehner, B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 2009, 138, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Davey, N.E.; Gibson, T.J. Proteome-wide analysis of human disease mutations in short linear motifs: Neglected players in cancer? Mol. Biosyst. 2014, 10, 2626–2642. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Kirchner, M.; Uyar, B.; Cheng, J.-Y.; Russo, G.; Hernandez-Miranda, L.R.; Szymborska, A.; Zauber, H.; Rudolph, I.-M.; Willnow, T.E.; et al. Mutations in Disordered Regions Can Cause Disease by Creating Dileucine Motifs. Cell 2018, 175, 239–253.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N.; The Cancer Genome Atlas Research Network; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nat. Cell Biol. 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.L.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nat. Cell Biol. 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Copeland, N.G.; Jenkins, N.A. Deciphering the genetic landscape of cancer—from genes to pathways. Trends Genet. 2009, 25, 455–462. [Google Scholar] [CrossRef]
- Ali, M.A.; Sjöblom, T. Molecular pathways in tumor progression: From discovery to functional understanding. Mol. BioSyst. 2009, 5, 902–908. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Petsalaki, E.; Rolland, T.; Hill, D.E.; Vidal, M.; Roth, F.P. Protein Domain-Level Landscape of Cancer-Type-Specific Somatic Mutations. PLoS Comput. Biol. 2015, 11, e1004147. [Google Scholar] [CrossRef] [Green Version]
- Tokheim, C.; Bhattacharya, R.; Niknafs, N.; Gygax, D.M.; Kim, R.; Ryan, M.; Masica, D.L.; Karchin, R. Exome-Scale Discovery of Hotspot Mutation Regions in Human Cancer Using 3D Protein Structure. Cancer Res. 2016, 76, 3719–3731. [Google Scholar] [CrossRef] [Green Version]
- Porta-Pardo, E.; Garcia-Alonso, L.; Hrabe, T.; Dopazo, J.; Godzik, A. A Pan-Cancer Catalogue of Cancer Driver Protein Interaction Interfaces. PLoS Comput. Biol. 2015, 11, e1004518. [Google Scholar] [CrossRef]
- Engin, H.B.; Kreisberg, J.F.; Carter, H. Structure-Based Analysis Reveals Cancer Missense Mutations Target Protein Interaction Interfaces. PLoS ONE 2016, 11, e0152929. [Google Scholar] [CrossRef]
- Kamburov, A.; Lawrence, M.S.; Polak, P.; Leshchiner, I.; Lage, K.; Golub, T.R.; Lander, E.S.; Getz, G. Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc. Natl. Acad. Sci. USA 2015, 112, E5486–E5495. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J. Cell regulation: Determined to signal discrete cooperation. Trends Biochem. Sci. 2009, 34, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Kumar, M.; Gibson, T.J.; Uyar, B.; Dosztányi, Z. Degrons in cancer. Sci. Signal. 2017, 10, eaak9982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichó, E.; Reményi, I.; Simon, I.; Mészáros, B. MFIB: A repository of protein complexes with mutual folding induced by binding. Bioinformatics 2017, 33, 3682–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Kimelman, D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J. Cell Sci. 2007, 120, 3337–3344. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.; Beare, D.; Bindal, N.; Bamford, S.; Ward, S.; Cole, C.; Jia, M.; Kok, C.; Boutselakis, H.; De, T.; et al. COSMIC: High-Resolution Cancer Genetics Using the Catalogue of Somatic Mutations in Cancer. Curr. Protoc. Hum. Genet. 2016, 91, 10–11. [Google Scholar] [CrossRef]
- Mészáros, B.; Zeke, A.; Reményi, A.; Simon, I.; Dosztányi, Z. Systematic analysis of somatic mutations driving cancer: Uncovering functional protein regions in disease development. Biol. Direct 2016, 11, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Buljan, M.; Blattmann, P.; Aebersold, R.; Boutros, M. Systematic characterization of pan-cancer mutation clusters. Mol. Syst. Biol. 2018, 14, e7974. [Google Scholar] [CrossRef]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Tabaro, F.; Mičetić, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidović, R.; Dosztányi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D1123–D1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef]
- Schad, E.; Fichó, E.; Pancsa, R.; Simon, I.; Dosztányi, Z.; Mészáros, B. DIBS: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2017, 34, 535–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, D.K.; Güntert, P. Objective identification of residue ranges for the superposition of protein structures. BMC Bioinform. 2011, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Mészáros, B.; Simon, I.; Dosztányi, Z. Prediction of Protein Binding Regions in Disordered Proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Mičetić, I.; Camilloni, C.; Davey, N.; Dosztányi, Z.; Mészáros, B.; Monzon, A.M.; et al. MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2017, 46, D471–D476. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonazzo, G.; Attrill, H.; Brown, N.; Marygold, S.J.; McQuilton, P.; Ponting, L.; Millburn, G.H. The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, H.; Gazit, E. The von Hippel-Lindau tumor suppressor protein is a molten globule under native conditions: Implications for its physiological activities. J. Biol. Chem. 2004, 279, 17190–17196. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J. Cell Sci. 2007, 120, 3327–3335. [Google Scholar] [CrossRef] [Green Version]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Garvie, C.W.; Hagman, J.; Wolberger, C. Structural Studies of Ets-1/Pax5 Complex Formation on DNA. Mol. Cell 2001, 8, 1267–1276. [Google Scholar] [CrossRef]
- Paz-Priel, I.; Friedman, A. C/EBPα dysregulation in AML and ALL. Crit. Rev. Oncog. 2011, 16, 93–102. [Google Scholar] [CrossRef]
- Hubbard, S.R. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hristova, K. Receptor tyrosine kinase transmembrane domains: Function, dimer structure and dimerization energetics. Cell Adh. Migr. 2010, 4, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Sangwan, V.; Park, M. Receptor Tyrosine Kinases: Role in Cancer Progression. Curr. Oncol. 2006, 13, 191–193. [Google Scholar] [CrossRef]
- Oppelt, P.J.; Hirbe, A.C.; Van Tine, B.A. Gastrointestinal stromal tumors (GISTs): Point mutations matter in management, a review. J. Gastrointest. Oncol. 2017, 8, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Deeb, K.K.; Smonskey, M.T.; DeFedericis, H.; Deeb, G.; Sait, S.N.; Wetzler, M.; Wang, E.S.; Starostik, P. Deletion and deletion/insertion mutations in the juxtamembrane domain of the FLT3 gene in adult acute myeloid leukemia. Leuk. Res. Rep. 2014, 3, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Pilotto, S.; Gkountakos, A.; Carbognin, L.; Scarpa, A.; Tortora, G.; Bria, E. MET exon 14 juxtamembrane splicing mutations: Clinical and therapeutical perspectives for cancer therapy. Ann. Transl. Med. 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Chase, A.; Schultheis, B.; Kreil, S.; Baxter, J.; Hidalgo-Curtis, C.; Jones, A.; Zhang, L.; Grand, F.H.; Melo, J.V.; Cross, N.C.P. Imatinib sensitivity as a consequence of a CSF1R-Y571D mutation and CSF1/CSF1R signaling abnormalities in the cell line GDM. Leukemia 2008, 23, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.L.; Holmes, K.A.; Carroll, J.S. FOXA1 mutations in hormone-dependent cancers. Front. Oncol. 2013, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, L.L.; Hambarchyan, A.; Meadows, J.D.; Barakat, N.H.; Pepa, P.A.; Breen, K.M.; Mellon, P.L.; Coss, D. Roles of Binding Elements, FOXL2 Domains, and Interactions With cJUN and SMADs in Regulation of FSHβ. Mol. Endocrinol. 2014, 28, 1640–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Antwerp, M.E.; Chen, D.G.; Chang, C.; Prochownik, E.V. A point mutation in the MyoD basic domain imparts c-Myc-like properties. Proc. Natl. Acad. Sci. USA 1992, 89, 9010–9014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Project, T.I.M.-S.; Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Lee, S.-Y.; Lee, J.; Jeong, C.-H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.-H.; Lee, H.J.; et al. USP8 Is a Novel Target for Overcoming Gefitinib Resistance in Lung Cancer. Clin. Cancer Res. 2013, 19, 3894–3904. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.L.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nat. Cell Biol. 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Perry, A.M.; Bierman, P.; Loberiza, F.; Nasr, M.R.; Szwajcer, D.; Del Bigio, M.R.; Smith, L.M.; Zhang, W.; Greiner, T.C. Frequency of MYD88 and CD79B mutations, and MGMT methylation in primary central nervous system diffuse large B-cell lymphoma. Neuropathology 2017, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-I.; Chen, C.-Y.; Lin, D.-T.; Tsay, W.; Tang, J.-L.; Yeh, Y.-C.; Shen, H.-L.; Su, F.-H.; Yao, M.; Huang, S.-Y.; et al. Characterization of CEBPA Mutations in Acute Myeloid Leukemia: Most Patients with CEBPA Mutations Have Biallelic Mutations and Show a Distinct Immunophenotype of the Leukemic Cells. Clin. Cancer Res. 2005, 11, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, S.A.; Worwood, M.; Oscier, D.; Jacobs, A.; Padua, R.A. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc. Natl. Acad. Sci. USA 1990, 87, 1377–1380. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Patel, L.; Mills, G.B.; Lu, K.H.; Sood, A.K.; Ding, L.; Kucherlapati, R.; Mardis, E.R.; Levine, D.A.; Shmulevich, I.; et al. Clinical Significance of CTNNB1 Mutation and Wnt Pathway Activation in Endometrioid Endometrial Carcinoma. J. Natl. Cancer Inst. 2014, 106, dju245. [Google Scholar] [CrossRef] [Green Version]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; Tone, A.A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod. Pathol. 2014, 27, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, J.T.; DeLaney, T.F.; Rosenberg, A.E.; Le, L.; Iafrate, A.J.; Kobayashi, W.; Szymonifka, J.; Yeap, B.Y.; Chen, Y.-L.; Harmon, D.C.; et al. β-Catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist 2013, 18, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Singh, V.; Verma, V.; Pandey, S.; Trivedi, R.; Singh, H.P.; Kumar, S.; Dwivedi, R.C.; Mishra, S.C. Current status and clinical association of beta-catenin with juvenile nasopharyngeal angiofibroma. J. Laryngol. Otol. 2016, 130, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Comino-Méndez, I.; De Cubas, A.A.; Bernal, C.; Álvarez-Escolá, C.; Sánchez-Malo, C.; Ramírez-Tortosa, C.L.; Pedrinaci, S.; Rapizzi, E.; Ercolino, T.; Bernini, G.; et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum. Mol. Genet. 2013, 22, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.; Butzow, R.; Andersson, N.; Alexiadis, M.; Unkila-Kallio, L.; Heikinheimo, M.; Fuller, P.J.; Anttonen, M. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod. Pathol. 2010, 23, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Köbel, M.; Senz, J.; Morin, R.D.; Clarke, B.A.; Wiegand, K.C.; Leung, G.; Zayed, A.; Mehl, E.; Kalloger, S.E.; et al. Mutation ofFOXL2in Granulosa-Cell Tumors of the Ovary. N. Engl. J. Med. 2009, 360, 2719–2729. [Google Scholar] [CrossRef]
- Gielen, G.H.; Gessi, M.; Hammes, J.; Kramm, C.M.; Waha, A.; Pietsch, T. H3F3A K27M mutation in pediatric CNS tumors: A marker for diffuse high-grade astrocytomas. Am. J. Clin. Pathol. 2013, 139, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Xu, Z.; Huo, X.; Tang, C.; Ye, H.; Nandakumar, V.; Lou, F.; Zhang, D.; Jiang, S.; Sun, H.; Dong, H.; et al. Frequent KIT Mutations in Human Gastrointestinal Stromal Tumors. Sci. Rep. 2015, 4, 5907. [Google Scholar] [CrossRef] [Green Version]
- Ravegnini, G.; Mariño-Enriquez, A.; Slater, J.; Eilers, G.; Wang, Y.; Zhu, M.; Nucci, M.R.; George, S.; Angelini, S.; Raut, C.P.; et al. MED12 mutations in leiomyosarcoma and extrauterine leiomyoma. Mod. Pathol. 2013, 26, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laé, M.; Gardrat, S.; Rondeau, S.; Richardot, C.; Caly, M.; Chemlali, W.; Vacher, S.; Couturier, J.; Mariani, O.; Terrier, P.; et al. MED12 mutations in breast phyllodes tumors: Evidence of temporal tumoral heterogeneity and identification of associated critical signaling pathways. Oncotarget 2016, 7, 84428–84438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, N.; Mehine, M.; Tolvanen, J.; Kaasinen, E.; Li, Y.; Lehtonen, H.J.; Gentile, M.; Yan, J.; Enge, M.; Taipale, M.; et al. MED12, the Mediator Complex Subunit 12 Gene, Is Mutated at High Frequency in Uterine Leiomyomas. Science 2011, 334, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Rekhi, B.; Upadhyay, P.; Ramteke, M.P.; Dutt, A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod. Pathol. 2016, 29, 1532–1540. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Huang, P.; Huang, X.; Li, X.; Feng, Z.; Li, F.; Liang, S.; Song, Y.; Stenvang, J.; Brünner, N.; et al. Comprehensive genomic analysis of Oesophageal Squamous Cell Carcinoma reveals clinical relevance. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Familiades, J.; Bousquet, M.; Lafage-Pochitaloff, M.; Béné, M.-C.; Beldjord, K.; De Vos, J.; Dastugue, N.; Coyaud, E.; Struski, S.; Quelen, C.; et al. PAX5 mutations occur frequently in adult B-cell progenitor acute lymphoblastic leukemia and PAX5 haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion genes: A GRAALL study. Leukemia 2009, 23, 1989–1998. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nat. Cell Biol. 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Ouyang, Y.; Qiao, C.; Chen, Y.; Zhang, S.-J. Clinical significance of CSF3R, SRSF2 and SETBP1 mutations in chronic neutrophilic leukemia and chronic myelomonocytic leukemia. Oncotarget 2017, 8, 20834–20841. [Google Scholar] [CrossRef]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2012, 45, 18–24. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Roller, A.; Haferlach, T.; Eder, C.; Dicker, F.; Grossmann, V.; Kohlmann, A.; Alpermann, T.; Yoshida, K.; Ogawa, S.; et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012, 120, 3080–3088. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, C.; Thiel, A.; Korah, H.E.; Reis, A.-C.; Saeger, W.; Stepanow, S.; Köhrer, K.; Reifenberger, G.; Knobbe-Thomsen, C.B.; Knappe, U.J.; et al. USP8 Mutations in Pituitary Cushing Adenomas—Targeted Analysis by Next-Generation Sequencing. J. Endocr. Soc. 2018, 2, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pajkos, M.; Zeke, A.; Dosztányi, Z. Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions. Biomolecules 2020, 10, 1115. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. On the relationship status for Arf and NPM1—It’s complicated. FEBS J. 2018, 285, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2008, 17, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.; Parker, J.P.; Marmion, C.J. Enzyme inhibition as a key target for the development of novel metal-based anti-cancer therapeutics. Anti-Cancer Agents Med. Chem. 2010, 10, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef]
- Lounnas, V.; Ritschel, T.; Kelder, J.; McGuire, R.; Bywater, R.P.; Foloppe, N. Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 2013, 5, e201302011. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, P. Intrinsically disordered proteins and prostate cancer: Pouring new wine in an old bottle. Asian J. Androl. 2016, 18, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [Green Version]
- Ballerini, P.; Struski, S.; Cresson, C.; Prade, N.; Toujani, S.; Deswarte, C.; Dobbelstein, S.; Petit, A.; Lapillonne, H.; Gautier, E.-F.; et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 2012, 26, 2384–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Ren, S.; Murphy, S.J.; Dalangood, S.; Chang, C.; Pang, X.; Cui, Y.; Wang, L.; Pan, Y.; Zhang, X.; et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol. Cell 2015, 59, 904–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesueur, F.; French Familial Melanoma Study Group; De Lichy, M.; Barrois, M.; Durand, G.; Bombled, J.; Avril, M.-F.; Chompret, A.; Boitier, F.; Lenoir, G.M.; et al. The contribution of large genomic deletions at the CDKN2A locus to the burden of familial melanoma. Br. J. Cancer 2008, 99, 364–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type (Name) | Implicated Gene Product | Malignancy | Incidence | Reference |
|---|---|---|---|---|
| Diffuse large B-cell lymphoma (ABC subtype) | CARD11 | malignant | 9.6–10.8% (7/73, 4/37) | [72,73] |
| Burkitt lymphoma | CCND3 | malignant | 14.6% (6/41) | [74] |
| Diffuse large B-cell lymphoma (ABC subtype) | CCND3 | malignant | 10.7% (3/28) | [74] |
| Diffuse large B-cell lymphoma (PCNS subtype) | CD79B | malignant | 31.6% (6/19) | [75] |
| Acute myeloid leukaemia | CEBPA | malignant | 15% (16/104) | [76] |
| Myelodysplasia and acute myeloblastic leukemia | CSF1R | malignant | 12.7% (14/110) | [77] |
| Endometrioid endometrial carcinoma (low-grade) | CTNNB1 | malignant | 87.0% (47/54) | [78] |
| Ovarian endometrioid carcinomas (low-grade) | CTNNB1 | malignant | 53.3% (16/30) | [79] |
| Hepatocellular carcinoma (HBV/HCV related) | CTNNB1 | malignant | 26% (32/122) | [80] |
| Desmoid tumor | CTNNB1 | benign | 73% (106/145) | [81] |
| Juvenile nasopharyngeal angiofibroma | CTNNB1 | benign | 75% (12/16) | [82] |
| Paraganglioma | EPAS1 | possibly malignant | 17% (7/41) | [83] |
| Adult granulosa cell tumors of the ovary | FOXL2 | malignant | 93–97% (52/56, 86/89) | [84,85] |
| Pediatric anaplastic astrocytoma/glioblastoma | H3F3A | malignant | 17.9–27.1% (5/28, 35/129) | [86] |
| Giant cell tumor of bone (stromal cell) | H3F3A | benign | 92% (49/53) | [87] |
| Chondroblastoma (stromal cell) | H3F3B | benign | 95% (73/77) | [87] |
| GIST | KIT | malignant | 47% (57/121) | [88] |
| Extrauterine leiomyoma and leiomyosarcoma | MED12 | (possibly) malignant | 19% (6/32) | [89] |
| Phyllodes tumor of breast | MED12 | possibly malignant | 49% (41/83) | [90] |
| Uterine leiomyoma | MED12 | benign | 70% (159/225) | [91] |
| Rhabdomyosarcoma | MYOD1 | malignant | 20% (10/49) | [92] |
| Esophageal squamous cell carcinoma | NFE2L2 | malignant | 9.6% (47/490) | [93] |
| B-cell progenitor acute lymphoblastic leukemia | PAX5 | malignant | 34–39% (40/117, 94/242) | [94,95] |
| Chronic myelomonocytic leukemia | SETBP1 | malignant | 25% (14/56) | [96] |
| Atypical Chronic Myeloid Leukemia | SETBP1 | malignant | 24.3% (17/70) | [97] |
| Chronic myelomonocytic leukaemia | SRSF2 | malignant | 47% (129/275) | [98] |
| Pituitary adenoma | USP8 | possibly malignant | 14% (6/42) | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mészáros, B.; Hajdu-Soltész, B.; Zeke, A.; Dosztányi, Z. Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules 2021, 11, 381. https://doi.org/10.3390/biom11030381
Mészáros B, Hajdu-Soltész B, Zeke A, Dosztányi Z. Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules. 2021; 11(3):381. https://doi.org/10.3390/biom11030381
Chicago/Turabian StyleMészáros, Bálint, Borbála Hajdu-Soltész, András Zeke, and Zsuzsanna Dosztányi. 2021. "Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies" Biomolecules 11, no. 3: 381. https://doi.org/10.3390/biom11030381
APA StyleMészáros, B., Hajdu-Soltész, B., Zeke, A., & Dosztányi, Z. (2021). Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules, 11(3), 381. https://doi.org/10.3390/biom11030381