
Human Cadaveric Donor Cornea Derived Extra Cellular Matrix Microparticles for Minimally Invasive Healing/Regeneration of Corneal Wounds


 ,
, 
Abstract
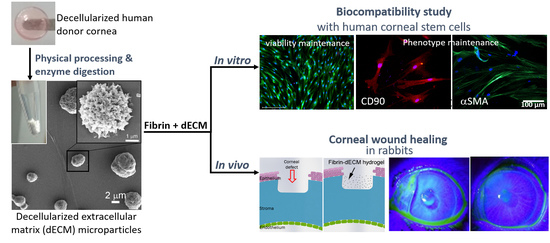
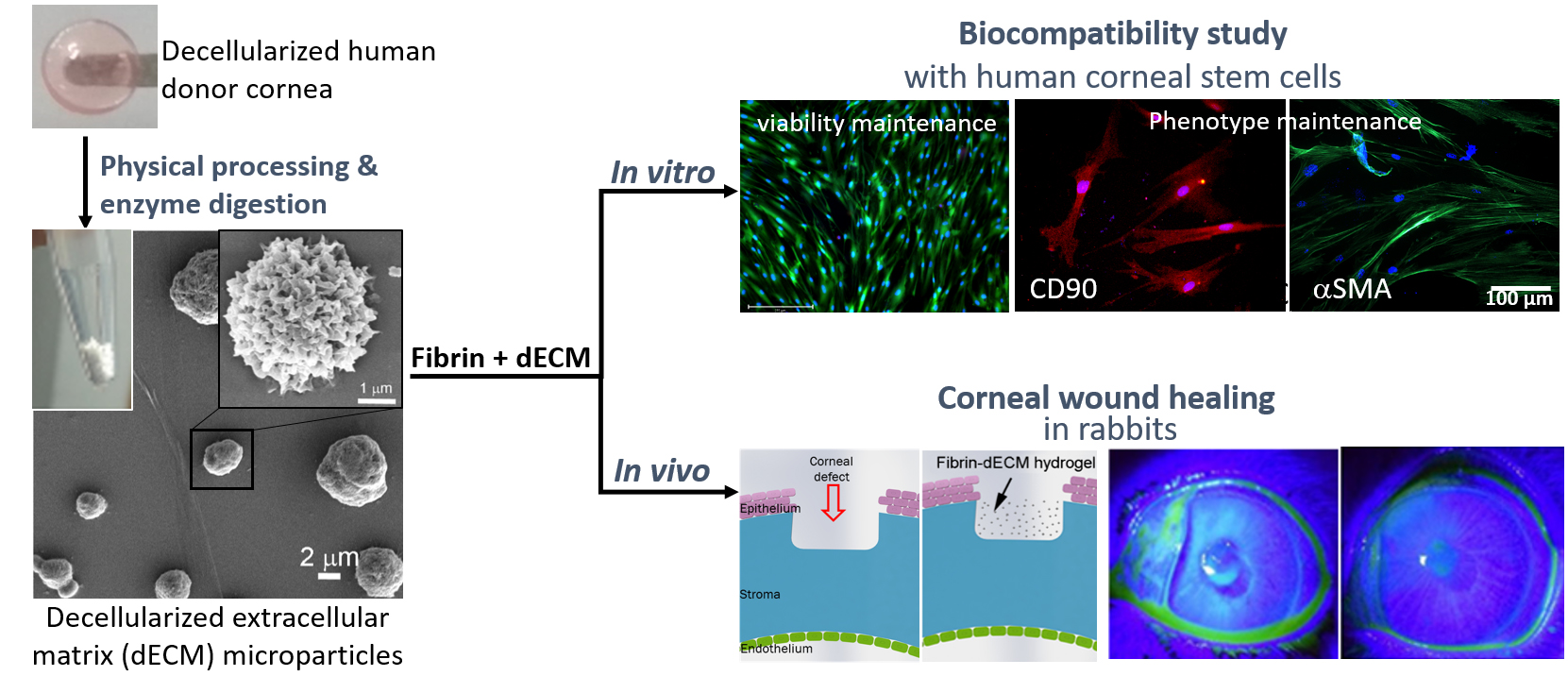
:
1. Introduction
2. Materials and Methods
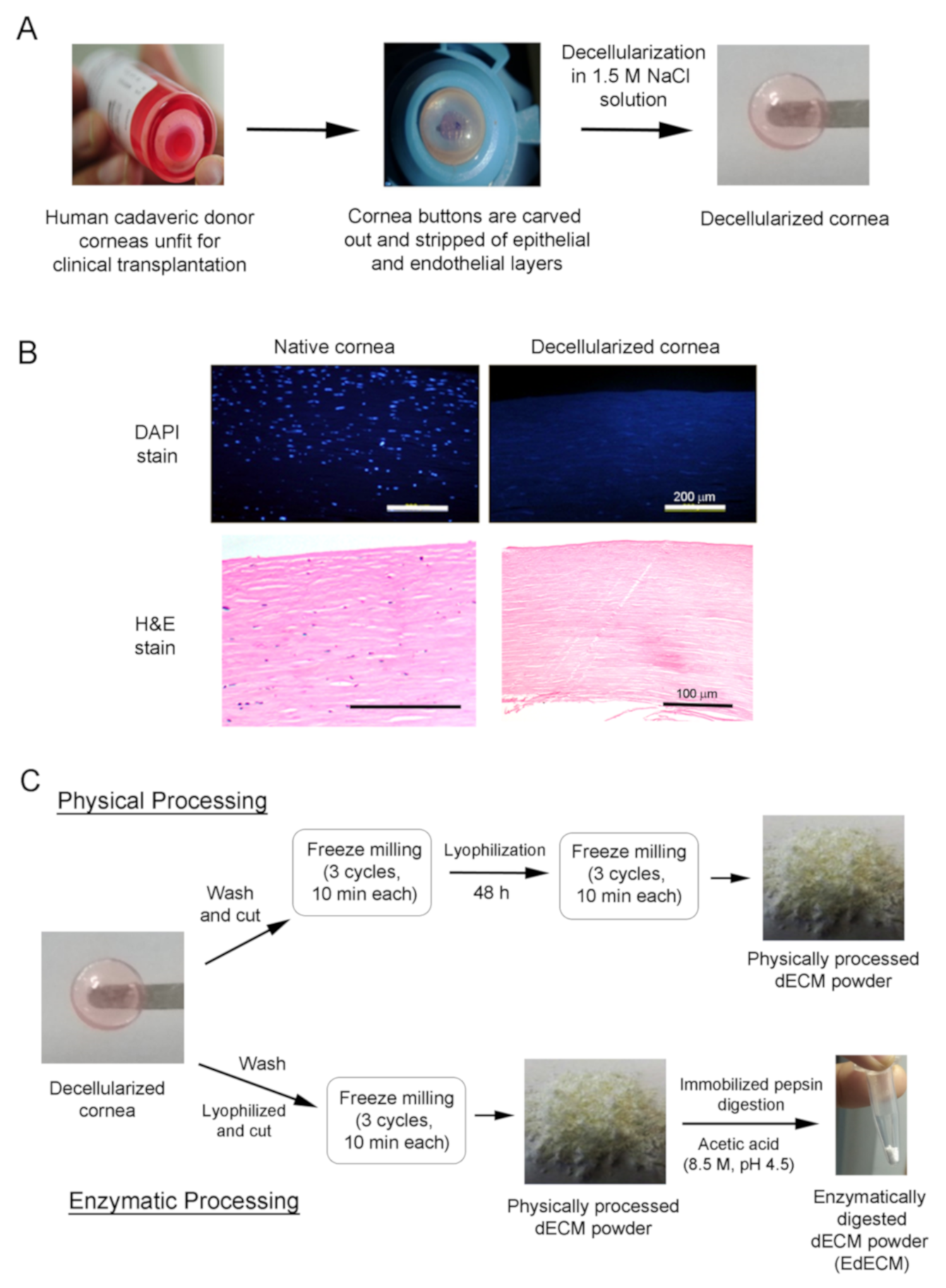
2.1. Decellularization of Human Cadaveric Donor Corneas
2.2. Processing of Decellularized Tissues
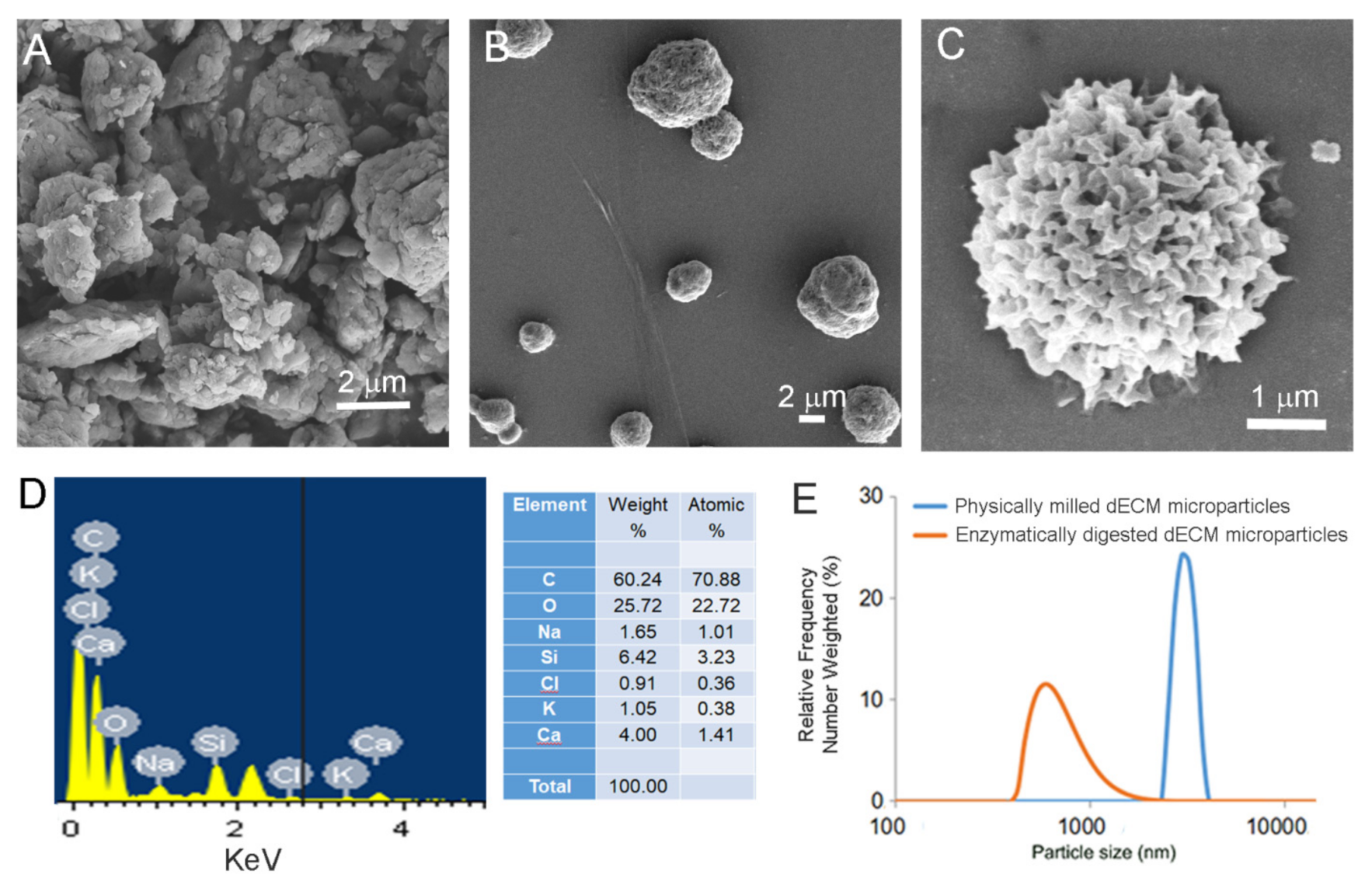
2.3. dECM Microparticle Characterization
2.3.1. Scanning Electron Microscopy (SEM) with Energy Dispersive X-Ray Analysis (EDAX)
2.3.2. Thermogravimetric Analysis (TGA)
2.3.3. Dynamic Light Scattering (DLS)
2.3.4. Fourier Transform Infrared (FTIR)
2.3.5. Sandwich ELISA Assay
2.3.6. Mass Spectrometry
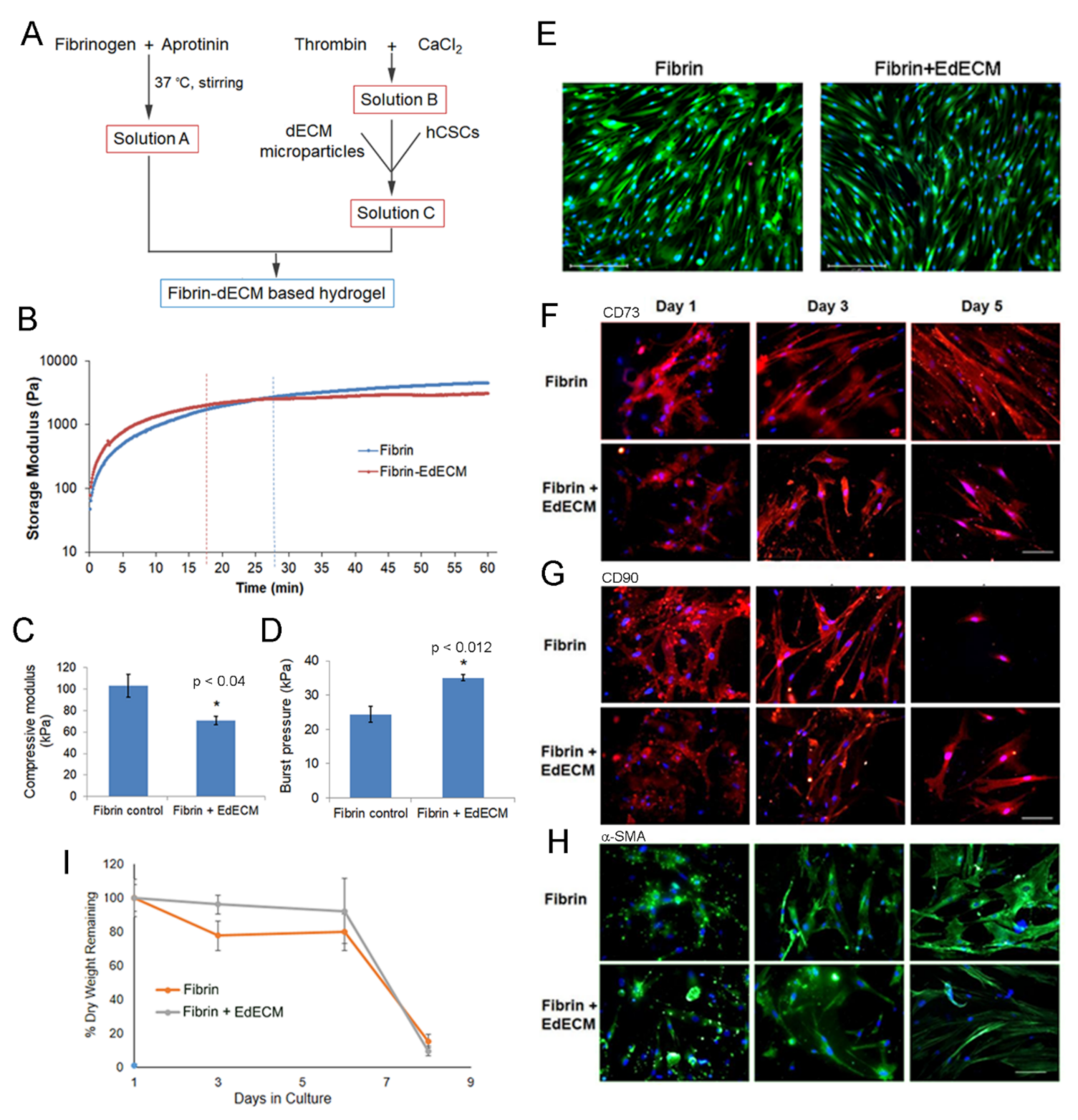
2.4. Hydrogel Preparation
2.5. Hydrogel Characterization
2.5.1. Compressive Modulus
2.5.2. Crosslinking Kinetics
2.5.3. Ex-Vivo Burst Pressure
2.5.4. MTT Assay on Hydrogel Extracts
2.5.5. Bacterial Reverse Mutation Test on Hydrogel Extracts
2.6. In Vitro Cell Culture Studies with hCSCS
2.6.1. Human Corneal Stem Cell Harvest and Expansion
2.6.2. Cell Encapsulation in Hydrogels
2.6.3. Live/Dead Assay
2.6.4. Biomarker Expression
2.6.5. Biodegradation Study In Vitro
2.7. In Vivo Studies
2.7.1. Skin Sensitization Test in Guinea Pigs
2.7.2. Acute Ocular Irritation Test in Rabbits
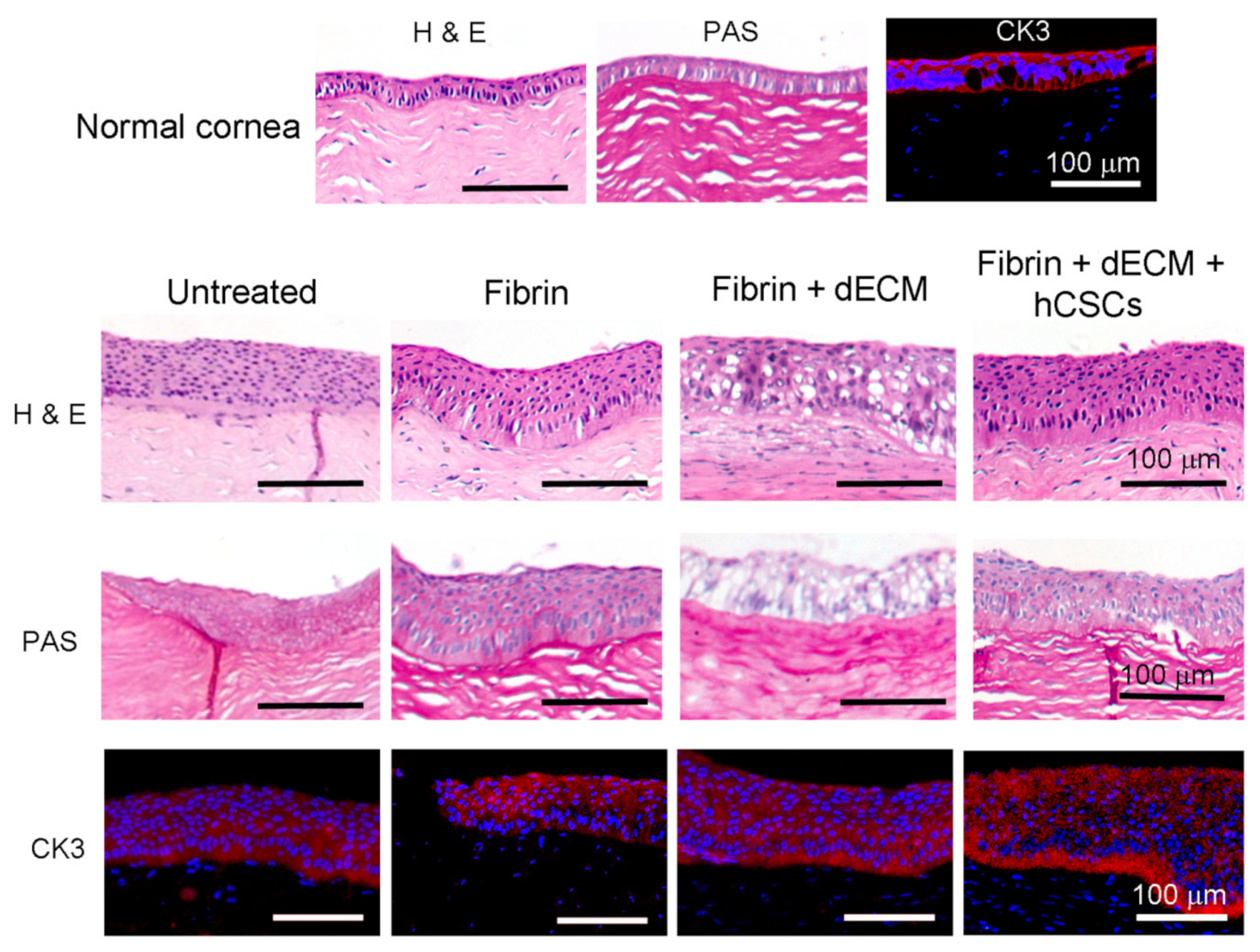
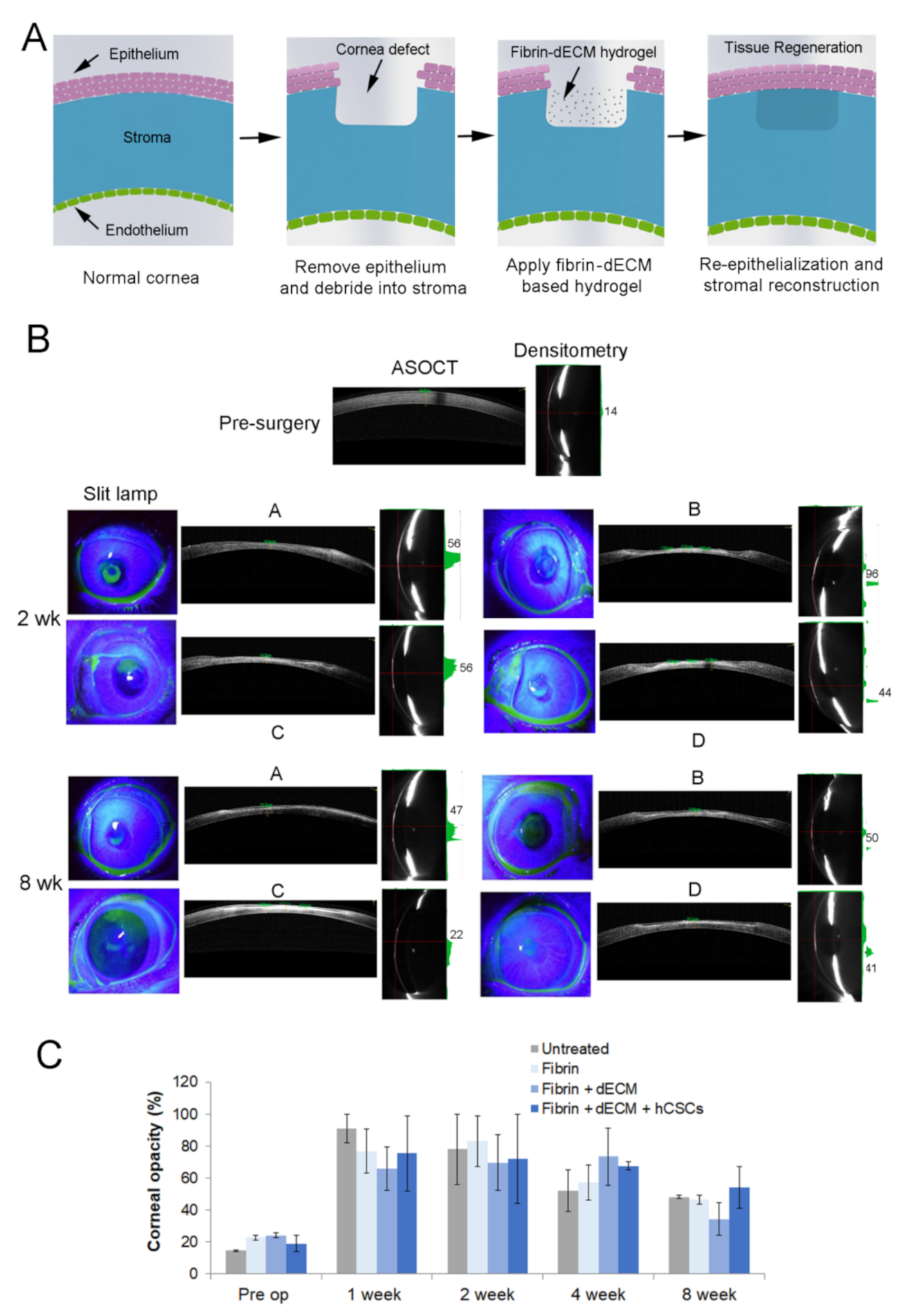
2.7.3. Treatment of Corneal Stromal Injury in Rabbit Model
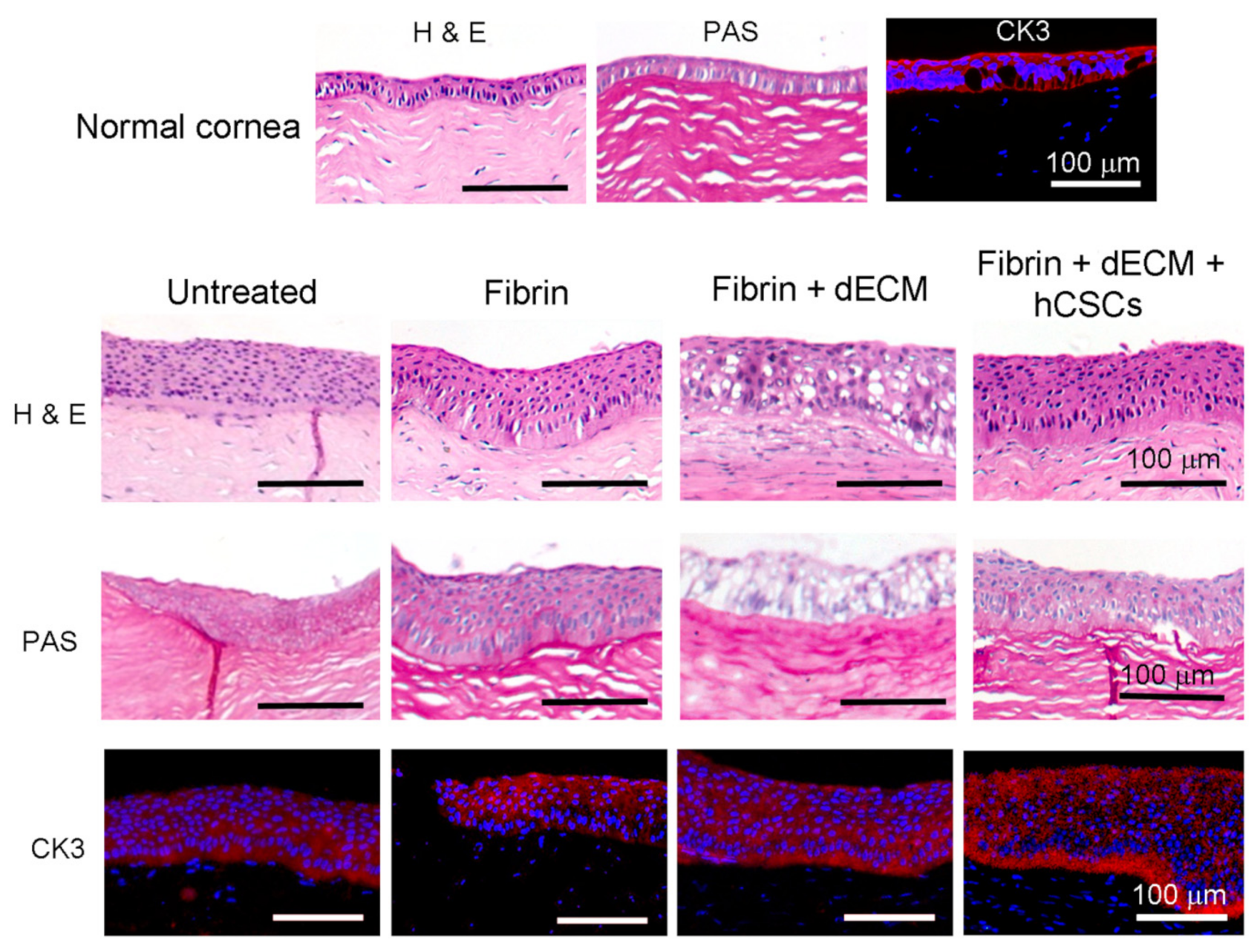
2.8. Histopathology
2.9. Immunofluorescence Staining
2.10. Statistical Analysis
3. Results and Discussion
3.1. Decellularization of Cadaveric Human Corneas
3.2. Characterization of Cornea Derived dECM Microparticles
3.3. Characterization of Fibrin and Fibrin–dECM Hydrogels
3.4. In Vitro hCSC Culture Studies
3.5. Animal Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, D.T.; Dart, J.K.; Holland, E.J.; Kinoshita, S. Corneal transplantation. Lancet 2012, 379, 1749–1761. [Google Scholar] [CrossRef]
- Stevens, G.A.; White, R.A.; Flaxman, S.R.; Price, H.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; Pesudovs, K.; Resnikoff, S.; et al. Global prevalence of vision impairment and blindness: Magnitude and temporal trends, 1990–2010. Ophthalmology 2013, 120, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Buznyk, O.; Reddy, J.C.; Pasyechnikova, N.; Alarcon, E.I.; Hayes, S.; Lewis, P.; Fagerholm, P.; He, C.; Iakymenko, S.; et al. Biomaterials-enabled cornea regeneration in patients at high risk for rejection of donor tissue transplantation. NPJ Regen. Med. 2018, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Trujillo-de Santiago, G.; Sharifi, R.; Yue, K.; Sani, E.S.; Kashaf, S.S.; Alvarez, M.M.; Leijten, J.; Khademhosseini, A.; Dana, R.; Annabi, N. Ocular adhesives: Design, chemistry, crosslinking mechanisms, and applications. Biomaterials 2019, 197, 345–367. [Google Scholar] [CrossRef] [Green Version]
- Jirásková, N.; Rozsival, P.; Burova, M.; Kalfertova, M. AlphaCor artificial cornea: Clinical outcome. Eye 2011, 25, 1138–1146. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Hertsenberg, A.J.; Funderburgh, M.L.; Burrow, M.K.; Mann, M.M.; Du, Y.; Lathrop, K.L.; Syed-Picard, F.N.; Adams, S.M.; Birk, D.E.; et al. Human limbal biopsy-derived stromal stem cells prevent corneal scarring. Sci. Transl. Med. 2014, 6, 266ra172. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Shukla, S.; Ramachandran, C.; Mishra, D.K.; Katikireddy, K.R.; Lal, I.; Chauhan, S.K.; Sangwan, V.S. Science and Art of Cell-Based Ocular Surface Regeneration. Int. Rev. Cell Mol. Biol. 2015, 319, 45–106. [Google Scholar] [CrossRef]
- Fagerholm, P.; Lagali, N.S.; Ong, J.A.; Merrett, K.; Jackson, W.B.; Polarek, J.W.; Suuronen, E.J.; Liu, Y.; Brunette, I.; Griffith, M. Stable corneal regeneration four years after implantation of a cell-free recombinant human collagen scaffold. Biomaterials 2014, 35, 2420–2427. [Google Scholar] [CrossRef] [Green Version]
- Badylak, S.; Kokini, K.; Tullius, B.; Simmons-Byrd, A.; Morff, R. Morphologic Study of Small Intestinal Submucosa as a Body Wall Repair Device. J. Surg. Res. 2002, 103, 190–202. [Google Scholar] [CrossRef]
- Gilbert, T.W.; Wognum, S.; Joyce, E.M.; Freytes, D.O.; Sacks, M.S.; Badylak, S.F. Collagen fiber alignment and biaxial mechanical behavior of porcine urinary bladder derived extracellular matrix. Biomaterials 2008, 29, 4775–4782. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Lu, Q.; Wang, X.; Majumdar, S.; Jun, A.S.; Stark, W.J.; Grant, M.P.; Elisseeff, J.H. Tissue-derived microparticles reduce inflammation and fibrosis in cornea wounds. Acta Biomater. 2019, 85, 192–202. [Google Scholar] [CrossRef]
- Kim, H.; Park, M.-N.; Kim, J.; Jang, J.; Kim, H.-K.; Cho, D.-W. Characterization of cornea-specific bioink: High transparency, improved in vivo safety. J. Tissue Eng. 2019, 10, 2041731418823382. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Wu, X.; Pang, K.; Yang, Y. Histological evaluation and biomechanical characterisation of an acellular porcine cornea scaffold. Br. J. Ophthalmol. 2010, 95, 410–414. [Google Scholar] [CrossRef]
- Márquez, S.P.; Martínez, V.S.; Ambrose, W.M.; Wang, J.; Gantxegui, N.G.; Schein, O.; Elisseeff, J. Decellularization of bovine corneas for tissue engineering applications. Acta Biomater. 2009, 5, 1839–1847. [Google Scholar] [CrossRef]
- Du, L.; Wu, X. Development and Characterization of a Full-Thickness Acellular Porcine Cornea Matrix for Tissue Engineering. Artif. Organs 2011, 35, 691–705. [Google Scholar] [CrossRef]
- Joshi, V.P.; Ojha, S.K.; Singh, V.; Basu, S. A reliable animal model of corneal stromal opacity: Development and validation using in vivo imaging. Ocul. Surf. 2020, 18, 681–688. [Google Scholar] [CrossRef]
- Wilson, S.L.; Sidney, L.E.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Corneal Decellularization: A Method of Recycling Unsuitable Donor Tissue for Clinical Translation? Curr. Eye Res. 2015, 41, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Andrades, M.; Cardona, J.D.L.C.; Ionescu, A.M.; Campos, A.; Perez, M.D.M.; Alaminos, M. Generation of Bioengineered Corneas with Decellularized Xenografts and Human Keratocytes. Investig. Opthalmol. Vis. Sci. 2011, 52, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Damala, M.; Swioklo, S.; Koduri, M.A.; Mitragotri, N.S.; Basu, S.; Connon, C.J.; Singh, V. Encapsulation of human limbus-derived stromal/mesenchymal stem cells for biological preservation and transportation in extreme Indian conditions for clinical use. Sci. Rep. 2019, 9, 16950. [Google Scholar] [CrossRef]
- Kethiri, A.R.; Raju, E.; Bokara, K.K.; Mishra, D.K.; Basu, S.; Rao, C.M.; Sangwan, V.S.; Singh, V. Inflammation, vascularization and goblet cell differences in LSCD: Validating animal models of corneal alkali burns. Exp. Eye Res. 2019, 185, 107665. [Google Scholar] [CrossRef]
- Coster, D.J.; Jessup, C.F.; Williams, K.A. Mechanisms of corneal allograft rejection and regional immunosuppression. Eye 2009, 23, 1894–1897. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Wang, D.-A. Decellularized orthopaedic tissue-engineered grafts: Biomaterial scaffolds synthesised by therapeutic cells. Biomater. Sci. 2018, 6, 2798–2811. [Google Scholar] [CrossRef]
- Breen, E.D.; Curley, J.G.; Overcashier, D.E.; Hsu, C.C.; Shire, S.J. Effect of Moisture on the Stability of a Lyophilized Humanized Monoclonal Antibody Formulation. Pharm. Res. 2001, 18, 1345–1353. [Google Scholar] [CrossRef]
- Nakamura, K.; Iwazawa, R.; Yoshioka, Y. Introduction to a new cell transplantation platform via recombinant peptide petaloid pieces and its application to islet transplantation with mesenchymal stem cells. Transpl. Int. 2016, 29, 1039–1050. [Google Scholar] [CrossRef] [Green Version]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta (BBA)—Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Rieder, E.; Steinacher-Nigisch, A.; Weigel, G. Human immune-cell response towards diverse xenogeneic and allogeneic decellularized biomaterials. Int. J. Surg. 2016, 36, 347–351. [Google Scholar] [CrossRef]
- Dyrlund, T.F.; Poulsen, E.T.; Scavenius, C.; Nikolajsen, C.L.; Thøgersen, I.B.; Vorum, H.; Enghild, J.J. Human Cornea Proteome: Identification and Quantitation of the Proteins of the Three Main Layers Including Epithelium, Stroma, and Endothelium. J. Proteome Res. 2012, 11, 4231–4239. [Google Scholar] [CrossRef]
- Pouliot, R.A.; Young, B.M.; Link, P.A.; Park, H.E.; Kahn, A.R.; Shankar, K.; Schneck, M.B.; Weiss, D.J.; Heise, R.L. Porcine Lung-Derived Extracellular Matrix Hydrogel Properties Are Dependent on Pepsin Digestion Time. Tissue Eng. Part C Methods 2020, 26, 332–346. [Google Scholar] [CrossRef]
- Fu, J. Strong and tough hydrogels crosslinked by multi-functional polymer colloids. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 1336–1350. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, Y.; Duan, L.; Wang, L.; Ren, X.; Gao, G. Enhancing the self-recovery and mechanical property of hydrogels by macromolecular microspheres with thermal and redox initiation systems. RSC Adv. 2017, 7, 16015–16021. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Garcia, M.A.; Sloan, S.R.; Nidenberg, B.; Khalifa, Y.M.; Buckley, M.R. Depth-Dependent Out-of-Plane Young’s Modulus of the Human Cornea. Curr. Eye Res. 2017, 43, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Sjøntoft, E.; Edmund, C. In vivo determination of young’s modulus for the human cornea. Bull. Math. Biol. 1987, 49, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, B.; Tahi, H.; Galand, A. Use of Human Fibrin Glue and Amniotic Membrane Transplant in Corneal Perforation. Cornea 2001, 20, 230–232. [Google Scholar] [CrossRef]
- Wang, Y.X.; Xu, L.; Bin Wei, W.; Jonas, J.B. Intraocular pressure and its normal range adjusted for ocular and systemic parameters. The Beijing Eye Study 2011. PLoS ONE 2018, 13, e0196926. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.-J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise Review: The Surface Markers and Identity of Human Mesenchymal Stem Cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- De Oliveira, R.C.; Wilson, S.E. Fibrocytes, Wound Healing, and Corneal Fibrosis. Investig. Ophthalmol. Vis. Sci. 2020, 61, 28. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.E. Corneal myofibroblast biology and pathobiology: Generation, persistence, and transparency. Exp. Eye Res. 2012, 99, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Meng, H.; Liu, Y.; Lee, B.P. Fibrin Gel as an Injectable Biodegradable Scaffold and Cell Carrier for Tissue Engineering. Sci. World J. 2015, 2015, 685690. [Google Scholar] [CrossRef]
- Sun, T.-T.; Tseng, S.C.G.; Huang, A.J.-W.; Cooper, D.; Schermer, A.; Lynch, M.H.; Weiss, R.; Eichner, R. Monoclonal Antibody Studies of Mammalian Epithelial Keratins: A Review. Ann. N. Y. Acad. Sci. 1985, 455, 307–329. [Google Scholar] [CrossRef]
- Chameettachal, S.; Prasad, D.; Parekh, Y.; Basu, S.; Singh, V.; Bokara, K.K.; Pati, F. Prevention of Corneal Myofi-broblastic Differentiation in vitro Using a Biomimetic ECM Hydrogel for Corneal Tissue Regeneration. ACS Appl. BioMater. 2021, 4, 533–544. [Google Scholar] [CrossRef]
- Gum, G.; Wirostko, B.; Rafii, M.; Pritt, S.; Gutierrez, D. Corneal Wound Healing Model in New Zealand White Rabbits for Evaluating Persistent Corneal Epithelial Defects. Investig. Opthalmol. Vis. Sci. 2013, 54, 3903. [Google Scholar]
- Chang, S.-W.; Chou, S.-F.; Chuang, J.-L. Mechanical corneal epithelium scraping and ethanol treatment up-regulate cytokine gene expression differently in rabbit cornea. J. Refract. Surg. 2008, 24, 150–159. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dECM Microparticle Characterization | |
|---|---|
| SEM | Physically processed dECM and EdECM |
| Thermogravimetric analysis (TGA) | Physically processed dECM |
| Dynamic Light Scattering (DLS) | Physically processed dECM and EdECM |
| Fourier Transform Infrared (FTIR) | Physically processed dECM and EdECM |
| Sandwich ELISA assay | EdECM |
| Mass Spectrometry | EdECM |
| Hydrogel Characterization | |
| Compressive modulus | EdECM |
| Crosslinking kinetics | EdECM |
| Ex-vivo burst pressure | EdECM |
| In vitro Cell Culture Studies with hCSCS | |
| MTT assay on hydrogel extracts | Physically processed dECM |
| Bacterial reverse mutation test on hydrogel extracts | Physically processed dECM |
| Cell encapsulation in hydrogels | EdECM |
| Live/Dead assay | EdECM |
| Biomarker expression | EdECM |
| Biodegradation Study in vitro | EdECM |
| In Vivo Studies | |
| Skin sensitization test in Guinea pigs | Physically processed dECM |
| Acute ocular irritation test in rabbits | Physically processed dECM |
| Treatment of corneal stromal injury in rabbit model | Physically processed dECM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandru, A.; Agrawal, P.; Ojha, S.K.; Selvakumar, K.; Shiva, V.K.; Gharat, T.; Selvam, S.; Thomas, M.B.; Damala, M.; Prasad, D.; et al. Human Cadaveric Donor Cornea Derived Extra Cellular Matrix Microparticles for Minimally Invasive Healing/Regeneration of Corneal Wounds. Biomolecules 2021, 11, 532. https://doi.org/10.3390/biom11040532
Chandru A, Agrawal P, Ojha SK, Selvakumar K, Shiva VK, Gharat T, Selvam S, Thomas MB, Damala M, Prasad D, et al. Human Cadaveric Donor Cornea Derived Extra Cellular Matrix Microparticles for Minimally Invasive Healing/Regeneration of Corneal Wounds. Biomolecules. 2021; 11(4):532. https://doi.org/10.3390/biom11040532
Chicago/Turabian StyleChandru, Arun, Parinita Agrawal, Sanjay Kumar Ojha, Kamalnath Selvakumar, Vaishnavi K. Shiva, Tanmay Gharat, Shivaram Selvam, Midhun Ben Thomas, Mukesh Damala, Deeksha Prasad, and et al. 2021. "Human Cadaveric Donor Cornea Derived Extra Cellular Matrix Microparticles for Minimally Invasive Healing/Regeneration of Corneal Wounds" Biomolecules 11, no. 4: 532. https://doi.org/10.3390/biom11040532
APA StyleChandru, A., Agrawal, P., Ojha, S. K., Selvakumar, K., Shiva, V. K., Gharat, T., Selvam, S., Thomas, M. B., Damala, M., Prasad, D., Basu, S., Bhowmick, T., Sangwan, V. S., & Singh, V. (2021). Human Cadaveric Donor Cornea Derived Extra Cellular Matrix Microparticles for Minimally Invasive Healing/Regeneration of Corneal Wounds. Biomolecules, 11(4), 532. https://doi.org/10.3390/biom11040532