2. Materials and Methods
2.1. Cord Blood CD34+ Cells
CD34
+ cells were isolated by Miltenyi’s indirect immunomagnetic procedure (Magnetic-activated cell sorting (MACS) human CD34
+ progenitor cell isolation kit, Miltenyi Biotec, Paris, France) with magnetic selection MS columns (Vario MACS device, Miltenyi Biotec, Paris, France) from the cord blood (CB) units noted as “not suitable for banking” at the CB Bank of Bordeaux, French Blood Institute. After selection, CD34
+ cells were either used directly for the experiments or were frozen for later use [
8].
2.2. CD34+ Expansion Model
CD34
+ cells from CB (2 × 10
5 cells/mL) were seeded with different concentrations of αTOA (0 µM, 20 µM, 40 µM, and 150 µM) in the culture medium supplemented with cytokines from Peprotech (Neuilly-Sur-Seine, France): SCF (Stem Cell Factor), (FMS-Like Tyrosine Kinase 3 receptor Ligand) Flt3L (100 ng/mL each) G-CSF (10 ng/mL), and TPO (20 ng/mL). The αTOA doses (20 µM and 40 µM) were chosen on the basis of plasma αTOC concentrations in human serum [
9,
10]. To comprehensively cover the αTOC effect, we also used a higher dose (150 µM) in order to evaluate αTOC toxicity. Owing to the difficulties in procuring the same medium throughout the study, a set of different media was used for all analyses (HP01 medium, Macopharma, Tourcoing, France, HPGM and X-VIVO 15 medium, Lonza, Basel, Switzerland) and the same effect of αTOA was observed with all the different media used.
In order to analyze energetic metabolism (oxygen consumption rate, glycolytic activity, ATP and ROS level) in the first step of expansion, CD34+ cells were cultured in the above-described conditions for only 24 h at 37 °C + 5% CO2 before analysis. For the other assays, CD34+ cells were cultured for 3 days in the same conditions prior to the analysis of the secondary culture.
After 3 days of incubation of the primary culture, cells were washed to remove αTOA and the secondary culture (2 × 10
4 cells/mL) was started in the same medium supplemented with the same cytokines. The primary/secondary culture approach is used to detect the activity of the primitive progenitors preceding colony forming cells (pre-CFC) [
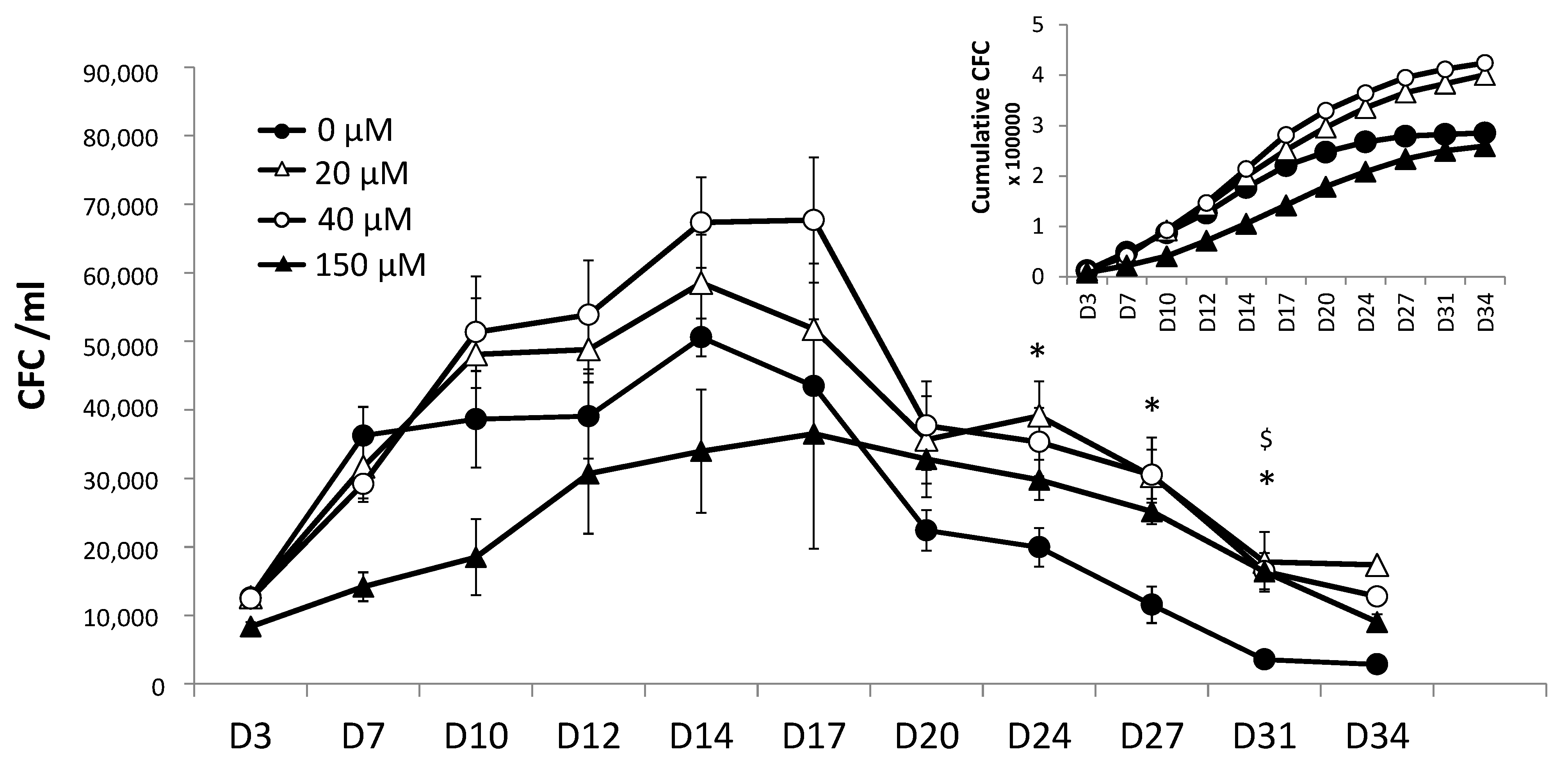
11]. We chose to compare different time-points in the kinetics of the expansion at the beginning and during the secondary culture: day 3, day 7, day 10, day 14, day 17, etc.
2.3. Colony-Forming Units Assay
For each analysis at day 0 (start of the primary culture), day 3 (end of the primary and start of the secondary culture), and for different time-points during the secondary culture, 50 cells were plated in cytokine-supplemented methylcellulose (H4034, stem cell technology, Grenoble, France) in 24-well plates in duplicate. After 14 days of incubation at 37 °C in a water-saturated atmosphere (20% O2 and 5% CO2), the colony-forming units (CFUs) were counted using an inverted microscope (Nikon, Champigny sur Marne, France).
2.4. Viability and Apoptosis Assay
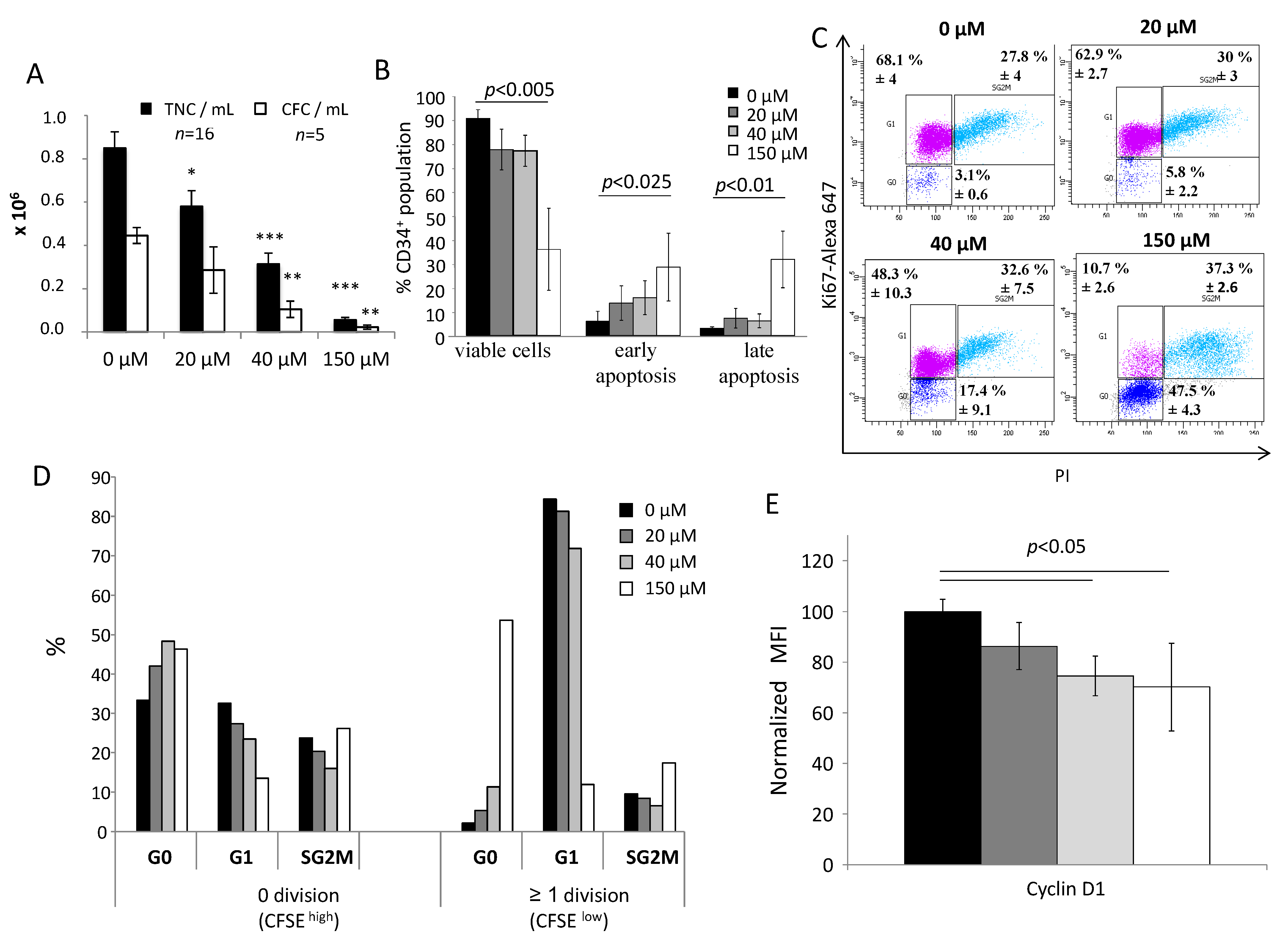
In order to investigate cell viability after 3 days of primary culture with αTOA at different concentrations, we used an Annexin V/propidium iodide (PI) kit (Beckman Coulter, Villepinte, France) paired with CD34+ immunolabelling (anti-human CD34-APC, Beckton Dickinson, Pont de Claix, France). Cells were analyzed with a BD FACS Canto II flow cytometer (Becton Dickinson, Rungis, France), after which we were able to evaluate 3 sub-populations: viable cells (Ann−, PI−), early apoptotic cells (Ann+, PI−) and apoptotic/necrotic cells (Ann+, PI+).
2.5. Cell Cycle Analysis
To analyze the quiescent state (G0 phase), G1 phase, or proliferative phases (S, G2, M) of the CD34+ cells after 3 days of primary culture with αTOA, we used Ki67/PI labelling after fixation and permeabilization steps. Cells were incubated overnight at 4 °C in phosphate-buffered saline (PBS) + 0.5% formaldehyde, then washed in PBS and incubated for a minimum of 2 h in 80% ethanol at −20 °C. Cells were washed in PBS and labelled with Alexa Fluor 647 anti-human Ki67 antibody (Beckton Dickinson, Pont de Claix, France) in a 0.25% Triton X-100 buffer for 1 h at 4 °C in the dark, then washed and resuspended in PBS–ribonuclease (1 mg/mL)–PI (50 mg/mL) buffer prior to analysis on a BD FACS Canto II flow cytometer.
In parallel, fixed and permeabilized cells were labelled with FITC anti-human cyclin D1 antibody set (#554109 BD Biosciences) for 1 h at 4 °C in the dark, then washed and resuspended in PBS prior to analysis on a BD FACS Canto II flow cytometer.
For the tracking of cell division, fresh CD34+ cells at 10 × 106 /mL were incubated on day 0 with 5 µM Carboxyfluorescein succinimidyl ester (CFSE) dye (#C34570, Thermo Fisher Scientific, Bordeaux, France) for 15 min at 37 °C and then washed twice with warm PBS. After 3 days of primary culture, cells were separated into two populations with a cell sorter (FACS ARIA, Becton Dickinson, Rungis, France) according to their fluorescence intensity. The CFSEhigh population (same fluorescence intensity as day 0), represents cells which did not undergo division, while CFSElow cells represent the population which had divided at least once. All cell populations were fixed and permeabilized for Ki67/PI labelling as described above.
2.6. Analysis of Primitive HSC Activity (Pre-SRCs)
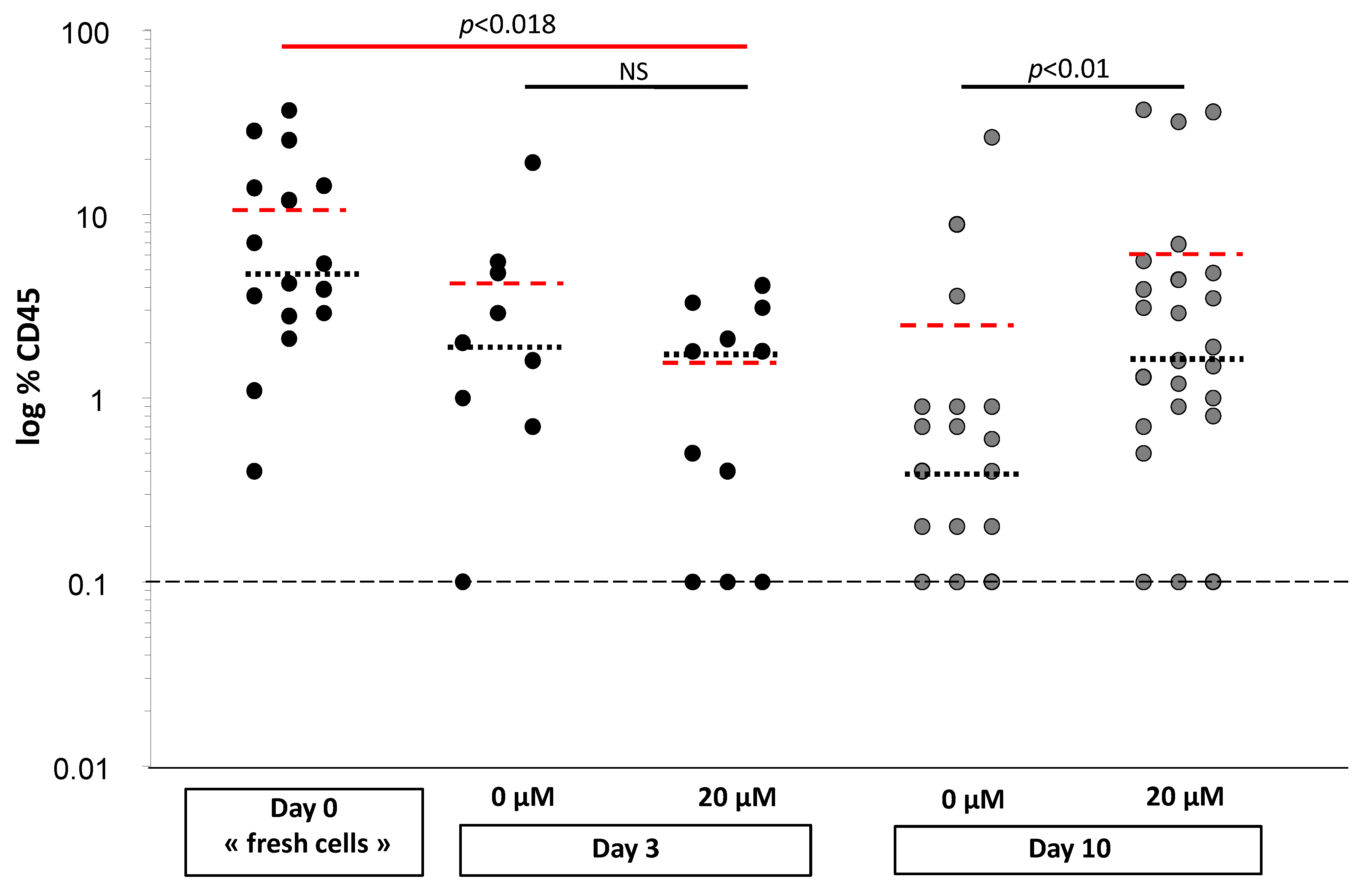
CD34+ cells treated with or without αTOA. After 3 days of primary expansion, the culture (PEC) was intravenously injected into 12-week-old NOD scid gamma mouse (NSG) mice (one part of the PEC), or washed and seeded in a secondary culture for 10 days (second part of the PEC), and then injected into a second group of mice. On day 0, 1000 cells were injected per mouse, or a number of cells representing their progeny (equivalent) were injected on day 3. On day 10, the equivalent of 1/10 of the day 0 cell progeny was injected. This reduction in the injected cell proportion was necessary in order to avoid saturation (high plateau chimerism) in the recipient mice, which can occur due to the expansion of reconstituting HSCs and would prevent the adequate comparison between 0 µM and 20 µM αTOA treatments.
Eight weeks after injection, bone marrow from the femurs of recipient mice was collected, counted and analyzed by flow cytometry (BD FACS Canto II, Rungis, France) for human CD45 chimerism (FITC anti-human CD45, #A07782, Beckman Coulter, Villepinte, France).
2.7. Analysis of Mitochondrial ROS (MitoSOX)
The level of mitochondrial ROS was quantified by the fluorescent probe MitoSOX (Thermo Fisher Scientific, M366008) in CD34+ cells treated with or without αTOC after 3 days of primary culture. To measure ROS produced by the mitochondria, the cells were washed and resuspended in Hanks’ Balanced Salt Solution (HBSS) (Thermo Fisher Scientific #14025092); then, 5 μM of MitoSOX was added and the cells were incubated for 15 min in the dark at 37 °C. Next, 50 µM verapamil was added in order to block the efflux pumps that reject the probe. After the incubations, cells were washed twice with PBS pre-warmed to 37 °C (Lonza, #17-516F, Basel, Switzerland). Intracellular fluorescence was detected by flow cytometry on a BD FACS Canto II and analyzed using FACSDiva software (Becton Dickinson, Rungis, France).
2.8. Metabolic Analysis
The measurement of energetic metabolism of CD34+ cells in each condition (with or without αTOC) was performed with Seahorse XF extracellular flux analyzers (Agilent technology, Santa Clara, CA, USA). We performed a mitochondrial stress test, glycolysis stress test and analysis of individual activity of complex I, complex II and complex IV. Before performing these tests, cells were glued in a reversible way with Cell-Tak (7 µg/cm2, Corning Life Science, Corning, NY, USA) and seeded just before analysis in the Seahorse plate at 2 × 105 cells per well in triplicate for each condition. Measurements were recorded for 133 min and analyzed using Microsoft Excel.
2.9. Mitochondrial Stress Test
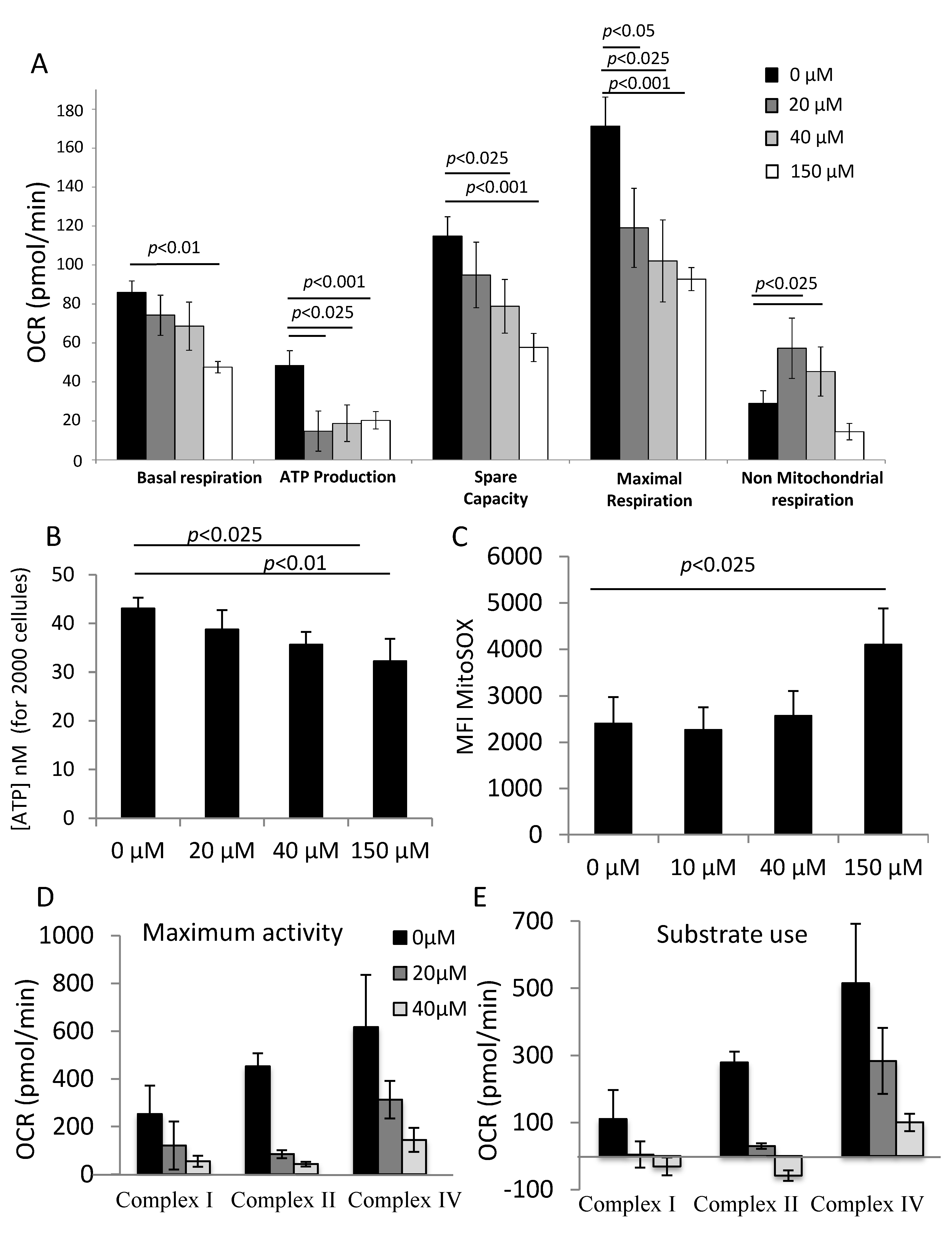
The mitochondrial stress test allows us to determine the basal respiration, ATP production, spare capacity, maximal respiration and non-mitochondrial oxygen consumption of CD34+ cells treated with or without αTOA. The cells were washed with PBS and the medium was replaced with pre-warmed XF Base Medium (part# 102353-100) supplemented with 100 mM pyruvate (Sigma Aldrich, St Louis, MO, USA, #P8574), 200 g/L glucose (Sigma Aldrich, #G8270) and 200 mM ultra-glutamine (Lonza, Basel, Switzerland, #BE04-684E), pH 7.4. The oxygen consumption rate (OCR) was recorded in real time before and after the subsequent injections of the modulatory compounds A, B, and C (A: 1.27 μM oligomycin (an ATP synthase inhibitor; #75351, Sigma Aldrich, USA), B: 100 μM of 2,4 Dinitrophenol (DNP) (a decoupling agent; #D198501 Sigma Aldrich, USA), and C: a mix of 1 μM rotenone (#R8875, Sigma Aldrich, USA) and 1 μM antimycin (#A8674 Sigma Aldrich, USA), which are inhibitors of complex I and complex III of the electron transport chain, respectively).
2.10. ATP Quantification by Luminescence
In order to correctly interpret ATP production, which was indirectly calculated from the Seahorse data, we chose to quantify ATP by a more sensitive method using the CellTiter-Glo® 2.0 Assay (G9241, Promega, Madison, WI, USA). This test provides a homogeneous method to determine the number of viable cells in culture by quantifying the amount of ATP present, which indicates the presence of metabolically active cells. We used ATP disodium salt (Cat.# P1132, Sigma Aldrich, Saint Louis, MO, USA) to generate the standard curve. The amount of ATP was determined after 24 h of culture on exactly 2000 CD34+ cells for all concentrations of αTOA tested (0 µM–150 µM).
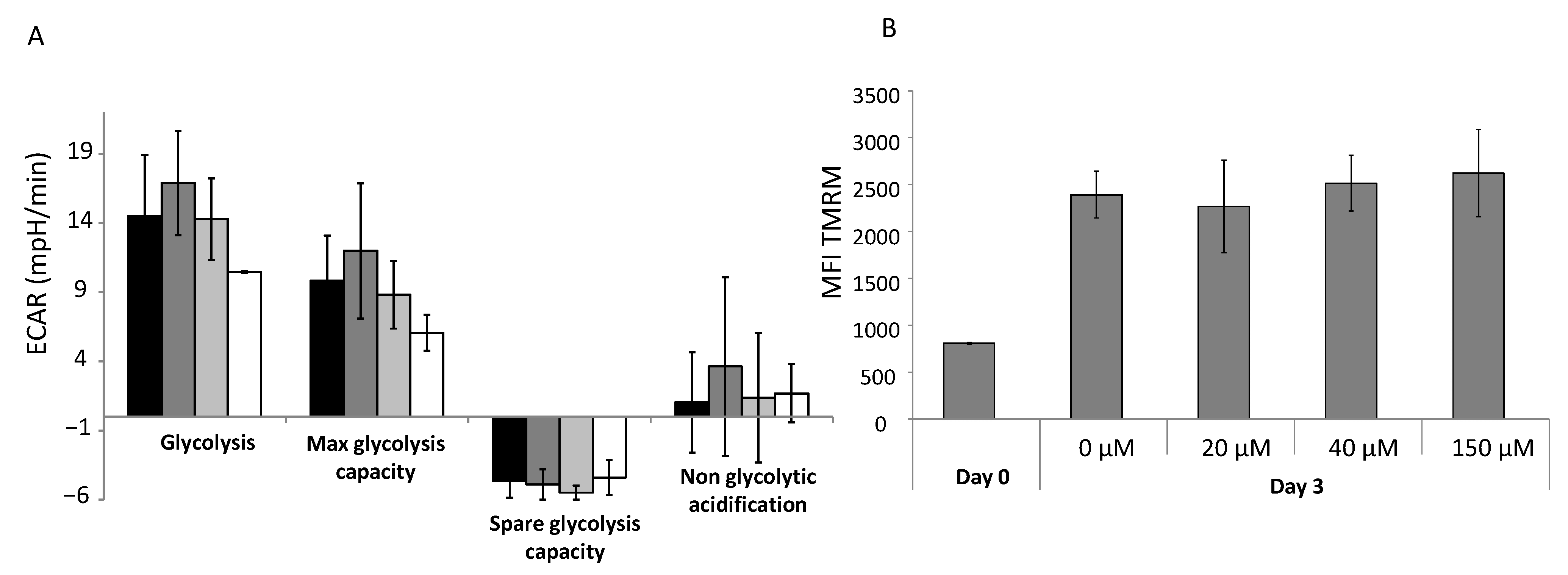
2.11. Glycolysis Stress Test
The glycolysis stress test, performed using Seahorse technology, allowed us to determine the basal glycolysis level, maximum/spare glycolytic capacity and non-glycolytic acidification. Cells were washed with PBS and the medium was replaced with pre-warmed XF Base Medium (part# 102353-100), pH 7.4. The extracellular acidification rate (ECAR) was recorded in real time before and after subsequent injections of the modulatory compounds A, B and C (A: 2 g/L glucose (#G8270, Sigma Aldrich, Saint Louis, MO), B: 1.27 μM oligomycin (#75351, Sigma Aldrich, Saint Louis, MO, USA), C: 100 mM 2–deoxy glucose, pH 7.4 (#D6134, Sigma Aldrich, Saint Louis, MO, USA)).
2.12. Analysis of Global Mitochondrial Activity
Global mitochondrial activity was quantified using the fluorescent probe tetramethylrhodamine methyl ester (TMRM; #M20036, Thermo Fisher Scientific, Waltham, MA, USA) in CD34+ cells treated with or without αTOA after 3 days of primary culture. The mitochondrial membrane potential state was determined directly by flow cytometry on the basis of a red–orange fluorescent signal emitted by TMRM (the signal diminishes with decreases in membrane potential). TMRM was added to 2 × 105 CD34+ cells (final concentration: 20 nM) treated with or without αTOA after 3 days in primary culture and incubated for 30 min in the dark at 37 °C. Then, 50 µM verapamil was added to each condition in order to block the efflux pumps that reject the probe. After the completed incubations, the cells were washed with cold PBS. Fluorescence was detected by a BD FACS Canto II flow cytometer using FACSDiva software. In further steps, cells were incubated with 100 µM DNP (2,4-dinitrophénol; Sigma Aldrich, St Louis, MO, USA) for 10 more minutes in the dark at 37 °C, washed, and the fluorescence was recorded for a second time. The difference between the levels of fluorescence before and after DNP was added corresponds to the specific signal of mitochondrial activity.
2.13. Individual Activity of Complex I, Complex II and Complex IV
To investigate how αTOA impacts the respiratory chain, we chose to apply the method described by Salabei et al. [
12] using the Seahorse device (Agilent Technologies, Santa Clara, CA, US). We individually analyzed the activity of respiratory chain complexes I, II and IV, as well as the rate of substrate use by each complex in permeabilized CD34
+ cells treated with or without αTOA for 24 h. On the day of the experiment, CD34
+ cells were washed with PBS and the culture medium was replaced with mannitol and sucrose buffer (MAS) pre-warmed at 37 °C, containing 70 mM sucrose (#573113, Calbiochim Sigma Aldrich), 220 mM mannitol (#A14030 Alfa Aesar by Thermo Fisher), 10 mM KH
2PO
4 (#529568, Calbiochim Sigma Aldrich), 5 mM MgCl
2 (#M8266, Sigma Aldrich), 2 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid #A14777 Alfa Aesar by Thermo Fisher) and 1 mM EGTA (Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid; E4378, Sigma Aldrich), pH 7.2. In this Seahorse protocol, we measured the oxygen consumption rate (OCR) of permeabilized cells treated with 25 μg/mL digitonin (# D141, Sigma Aldrich) in real time. First, we injected the specific substrate of the analyzed complex (2 mM malate (#M0875, Sigma Aldrich) for complex I, 10 mM succinate (#S3674, Sigma Aldrich) for complex II, and ascorbate (#A 4544, Sigma Aldrich) for complex IV). Next followed the injection of 1.27 μM olygomycin (#75351, Sigma Aldrich) and finally the injections of the specific inhibitors for each tested complex (10 μM rotenone (#R8875, Sigma Aldrich) for complex I, 10 μM antimycin (#A8674, Sigma Aldrich) for complex II, and Azide K (#740411, Sigma Aldrich) for complex IV) [
12].
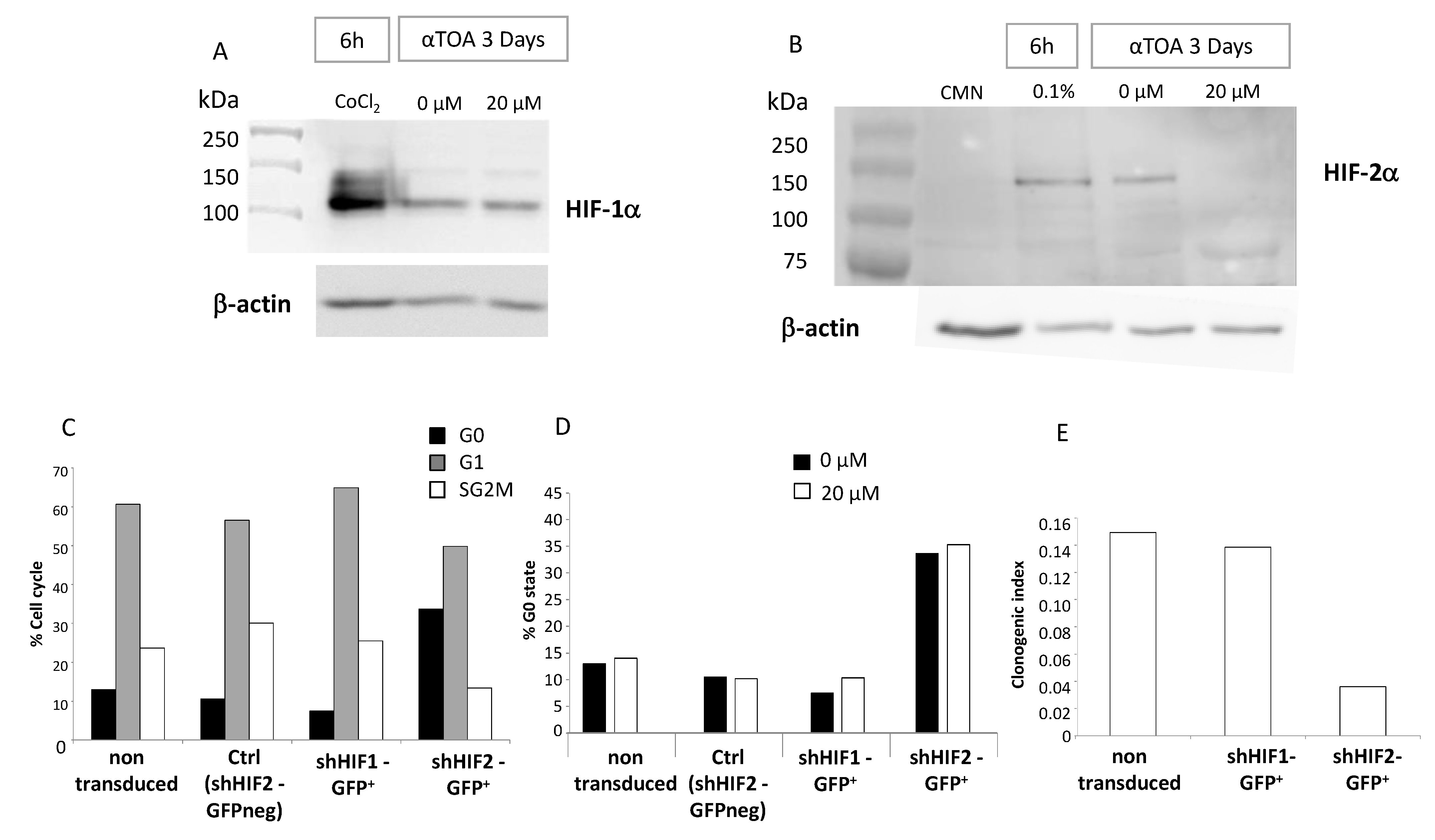
2.14. Western Blot of HIF-α Proteins
Proteins were extracted from CD34+ cells exposed to 0 μM, 20 μM and 40 μM of αTOA for 72 h at 20% O2 using cell lysis buffer (#9803, Cell Signaling Technology, Danvers, MA, USA). For the positive control, CD34+ cells were exposed to 100 μM cobalt chloride solution (CoCl2; #15862, Sigma Aldrich) or hypoxic conditions (0.1% O2) for 6 h in the same culture conditions as above, and total proteins were obtained using the same lysis buffer. For the negative control, mononucleated cells (MNCs) from CB were extracted on day 0 and total proteins were obtained using the same lysis buffer. Precision Plus Protein™ Dual Color Standards (#1610374, BioRad, Hercules, CA, USA) were used as protein markers.
Western blot analyses were performed with 30 µg of the total protein extract per condition (with/without αTOA). The protein extracts were separated according to molecular weight using ready-to-use 8–16% Mini-PROTEAN® TGX™ Precast Protein Gels (#4561103, BioRad, Hercules, CA, USA) in a BioRad electrophoresis system (power at 25 mA (per gel) for 90 min).
The separated proteins were transferred onto Hydrophilic polyvinylidene fluoride (PVDF) membrane (#1620177, BioRad, Hercules, CA, USA) with a wet transfer system for 2 h at 300 mA with FlashBlot transfer buffer (R-03090-D50, Advansta, San Jose, CA, USA). The membranes were blocked with Tris buffered saline with 0.1% Tween-20 (TBS–T) supplemented with 5% fat dried milk for 1 h at room temperature under agitation. After saturation, the membranes were incubated with 1:1000 HIF-1α (Abcam, #ab51608, Cambridge, UK), 1:500 HIF-2α (#NB100-132, Novus Biologicals, Bio-Techne, Centennial, CO, USA), or with 1: 5000 β-actin (#8226, Abcam, Cambridge, UK) primary antibody overnight at 4 °C under soft agitation. The blots were washed 3 times with 1× TBS-T at room temperature and then incubated with horseradish peroxidase-conjugated rabbit (R-05072-500, Advansta, San Jose, CA, USA) or mouse (R-05071-500, Advansta, San Jose, CA, USA) secondary antibody for 1 h at room temperature. Then, the membranes were washed 3 times and the immunoreactive proteins were detected by using the WesternBright Sirius Chemiluminescent Detection Kit (#K-12043-D10, Advansta, San Jose, CA, USA) and a Fusion Solo S6 edge device (Vilber Lourmat, France). Quantum software was used for the band intensity analysis (Vilber Lourmat, France).
2.15. qPCR
At the same time as protein extraction, some CD34+ cells were used to isolate total RNA using TRIzol reagent (#15596026 Invitrogen, Villebon-sur-Yvette, France), and reverse transcription was performed using a QuantiNova™ Reverse Transcription Kit (#205410, Quiagen, Courtaboeuf, France). The purity and quantity of the extracted RNA were verified with a Nanodrop device (Thermo Fisher Scientific, Waltham, MA, USA). qPCR was performed using Master Mix Applied Biosystems™ PowerUp™ SYBR™ Green (#A25780, Thermo Fisher Scientific) in a 384-well plate thermocycler (CFX 384 Touch PCR real-time detection system Bio-Rad, Hercules, CA, USA). The sequences of the forward/reverse HIF-α primers used are GCCCAATAGCCCTGAAGACT/TGAAATCCGTCTGGGTACTGC, which were validated with good efficacy (>95%).
2.16. Transduction Model of shHIF-α
In order to better understand the effect of αTOA on HIF-2α, we induced HIF-1α and HIF-2α shutdown via the lentiviral transduction of CD34+ cells. All lentivirus vectors were produced using the vectorology platform Vect’UB (INSERM US 005—CNRS 3427—TBMCore, Bordeaux University, France). CD34+ cells were transduced for 3 days with small hairpin RNA against HIF-1α (shHIF-1α) and/or against HIF-2α (shHIF-2α) for each experimental condition, and with shCTRL for the control group in Iscove Modified Dulbecco Media (IMDM) (Gibco), 50% BIT9500 (StemCell Technologies, Saint-Egrève, France) and the cytokines (Peprotech) SCF (100 ng/mL), FLT3-L (100 ng/mL), IL-6 (60 ng/mL) and TPO (20 ng/mL). Cells were seeded at a concentration of 1 × 106 cell/mL and transduced at 50 Multiplicity of infection (MOI) (25 MOI added on day 1 and 25 MOI added on day 2). After 3 days of transduction, cells were washed twice with PBS and GFP positive cells were sorted (BD FACS Aria, Beckton Dickinson, Rungis, France) to select only the transduced cells. GFP negative cells were kept for controls as non-transduced cells.
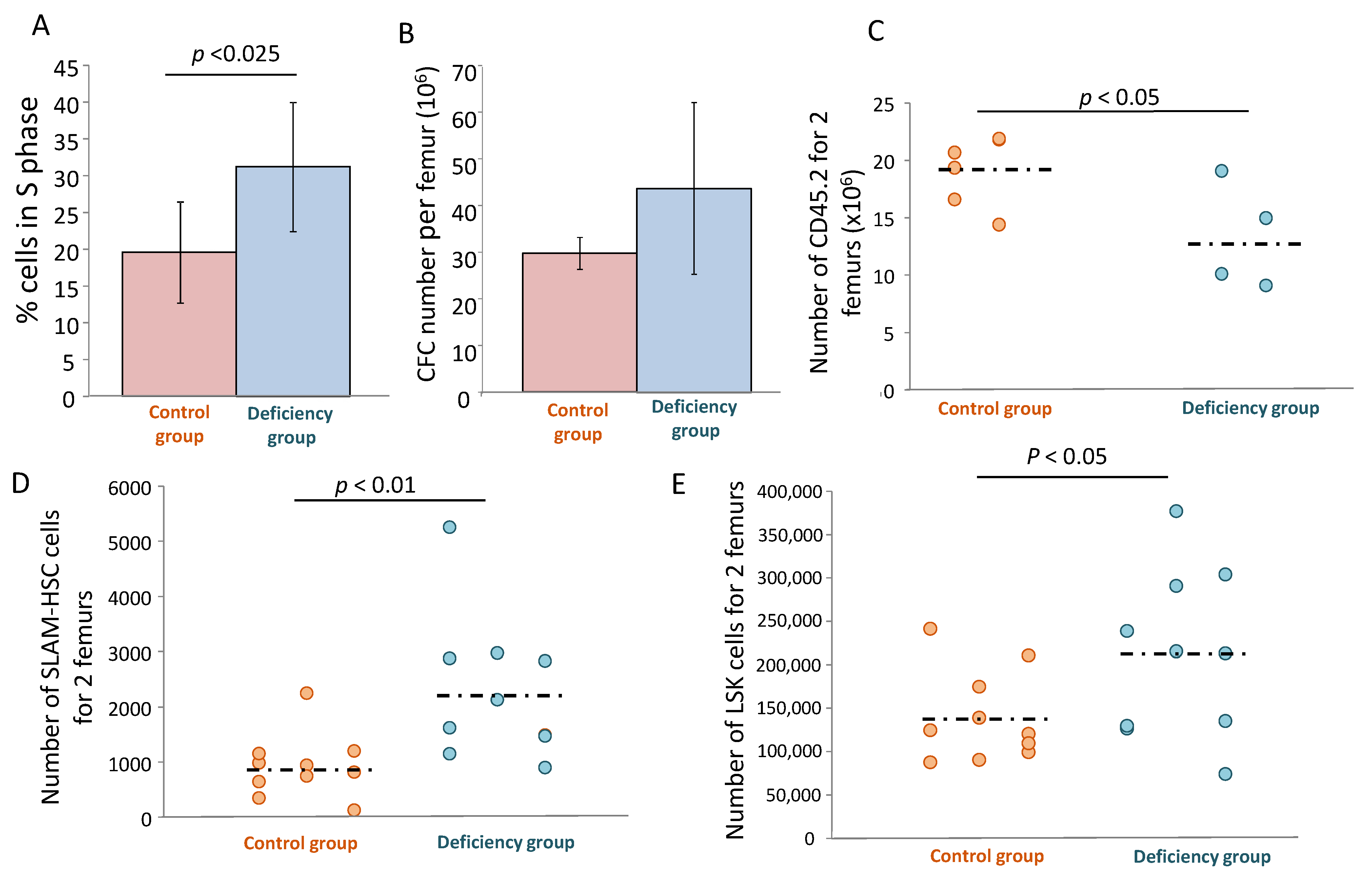
2.17. Murine αTOC Deficiency Model
These experiments were performed after approval from the Bordeaux Ethics Committee for animal experimentation (French National Ministry of High Education and Research authorization (APAFIS#13468-2018020914295792v4)). Three-week-old C57BL/6 CD45.2 mice (Charles River, Écully, France) were divided into 2 groups: (1) mice receiving vitamin E-free food (custom food U8959, Safe nutrition, Augy, France) (“deficiency” group), and (2) mice receiving the same U8959 food supplemented with 40 mg/kg of αTOA (“control” group). αTOC deficiency was validated after 2 months by quantification of the αTOC levels in mouse serum using HPLC (subcontracted by Eurofins Biomnis, Ivry-sur-Seine, France). After 2 months, the mice were sacrificed and the total number of cells, CFC, LSK and SLAM compartments were determined in their femoral bone marrow. The LSK and SLAM compartment were evaluated by flow cytometry. Bone marrow cells were incubated with PE anti-mouse CD3 (#100205), PE anti-mouse CD4 (#100407), PE anti-mouse CD8a (#100707), PE anti-mouse CD11b (#101207), PE anti-mouse B220 (#103207), PE anti-mouse Ly-6G (#108407), PE anti-mouse Ter119 (#116207), FITC anti-mouse CD117 (#105805) and APC anti-mouse Sca1 (#108111) antibodies for LSK determination, and BV421 anti-mouse CD48 (#103427) and BV510 anti-mouse CD150 (#115929) antibodies for SLAM determination. All antibodies were provided by BioLegend. S phase quantification was determined by the Ara-C suicide kit (cytosine-arabinoside; Cytarabine, #C3350000, Merck, Darmstadt, Germany). Femoral bone marrow (BM) cells were incubated for 1 h at 37 °C with or without Ara-C (50 µg/mL) and washed twice before being seeded (20,000 cells) in 1 mL of methylcellulose (R&D system, HSC007, Minneapolis, MN) and placed in a 35 mm petri dish in duplicate. After 7 days, the colonies were counted in both conditions. The percentage of reduction in the colony number in the Ara-C treated condition corresponds to the percentage of HPCs in the S phase of the cell cycle. To test their engraftment capacity, 100,000 cells from each donor mouse (5 mice in each group) were injected into a single C57BL/6 CD45.1 recipient mouse (i.v.) (Charles River). Four months after injection, recipient mice were sacrificed and their bone marrow cells were extracted and incubated with Alexa Fluor 700 anti-mouse CD45.1 (#110723, BioLegend), PE/Cy7 anti-mouse CD45.2 (#109829, BioLegend) and LSK-SLAM antibodies as described above. Cells were analyzed by flow cytometry (BD FACS Canto II) to determine the percentage of CD45.2 donor cells and to evaluate the LSK-SLAM compartment.
2.18. Statistical Analysis
Data from each cohort are presented as mean +/− standard deviation. The non-parametric Mann–Whitney test was used to compare experimental conditions for both in vitro and in vivo assays when two cohorts were compared. For the comparison of multiple cohorts, the Kruskal–Wallis test followed by the Mann–Whitney test with Bonferroni correction was used.
4. Discussion
In previous studies, it was concluded that αTOC (succinate or hemisuccinate), an αTOC derivative, does not have the same effect on normal hematopoietic cells as it does on leukemic cells [
13,
14]. However, to the best of our knowledge, the effect of αTOC on non-differentiated hematopoietic cells exhibiting the properties of HSPC was neither thoroughly nor rigorously investigated up until now. We investigated another αTOC analog, αTOA, which can be directly used in clinical-grade procedures. The results presented in this study show that αTOA has a toxic effect on the cells when administered in high, supra-physiological doses (150 µM), while this effect is absent at physiologically relevant doses (20 µM and 40 µM).
The ex vivo data obtained from the CB CD34
+ cells show that αTOA exhibits multiple effects on HSPCs, leading to increases in the HSPC proportion “arrested” in the quiescent state (G0 phase), as well as improved maintenance of their proliferative capacity. This latter effect may be relevant for HSC and HPC ex vivo engineering, particularly for the protocols where expansion should be combined with a “purge” that targets actively cycling cells. Alternatively, this approach might be used in ex vivo protocols for the amplification of the HPC fraction to prevent exhaustion of HSCs. We show that αTOA attenuates mitochondrial OXPHOS; however, the main mechanism underlying the accumulation in the G0 phase appears to be related to the inhibition of HIF-2α expression by αTOA. Although more thorough research is necessary to reveal the exact link between the attenuation of OXPHOS and
HIF-2α expression, an increase in ROS (detected after 72 h in our case) cannot be discarded. The increase in ROS is expected when real attenuation of OXPHOS occurs, such as when applied HIF-2α causes a decrease in ATP levels in a dose-dependent manner. Our results point to a blockage of the respiratory chain complexes I and II, which prompted us to hypothesize that αTOA may compete for electrons with ubiquinone/ubiquinol (coenzyme Q10 (CoQ10)), a molecule of very similar structure [
15,
16] that is critical for electron transport activity. αTOA may, in competition with CoQ10, undergo cyclic oxidation–reduction during the oxidation of substrates in the citric acid cycle, i.e., the reduction of fumarate to succinate. This hypothesis remains to be further explored, as well as the one concerning the effect of αTOA on the metabolism of lipids and amino acids in order to explain the sustained mitochondrial activity (ongoing work).
It is interesting to note that another αTOC supplement, α-tocopheryl succinate (αTOS), induces apoptosis in cancerous and leukemic cells via the blockage of either complex I [
17] or complex II activity by interacting with the proximal and distal ubiquinone-binding sites [
18].
Based on the data presented in this study, we conclude that αTOA does not affect CD34
+ cells in the active phases of the cell cycle, but rather prevents their exit from the G0 phase. When applied at the non-physiological, toxic dose of 150 µM, αTOA caused re-entrance of the cells into the G0 phase. This effect was shown for CB CD34
+ cells stimulated by IL-3 in response to an extremely low O
2 concentration [
11]; however, in the case of αTOA, it cannot be regarded as relevant due to the toxicity of the applied dose. It is possible that the higher proliferative capacity of the cells cultured in the presence of αTOA, revealed by an enhanced repopulation of the secondary cultures by HPC (CFC) and HSC (SRC), simply results from a better preserved quiescent fraction of the HSC. This is why the “HIF-2α issue” emerges as the most interesting point of this study. Treatment with αTOA abrogates HIF-2α expression (starting from the RNA level (
Figure S3)) and induces an accumulation of CD34
+ cells in the G0 phase. This G0 accumulation is also induced by shHIF2-α treatment, but it is not enhanced if αTOA is added simultaneously with shHIF-2α. The absence of an additive or synergic effect suggests that both shHIF2-α and αTOA target the same mechanism. The data presented here clearly show that the silencing of HIF-2α synthesis leads to an increase in the G0 proportion of human CD34
+ cells and, simultaneously, to the better maintenance of primitive HSC proliferative capacity/clonogenic potential. However, one cannot claim that HIF-2α is not a positive factor in HSC maintenance on the basis of these data obtained with CD34
+ cells. In fact, real HSC are a minority inside the CD34
+ cell population. Thus, we cannot question the result of the previous study with human cells [
19]. The results in mice showed that the positive in vivo effect of HIF-2α on the maintenance of hematopoiesis is associated with the functional bone marrow microenvironment [
20], while HIF-2α is not relevant for the maintenance of the HSCs themselves [
21]. Hence, in regard to this observation, it has been considered that HIF-2α regulatory competencies may be different in human and in mouse [
22]. The cellular and molecular responses depend on a cocktail of cytokines and other conditions of culture, so a more thorough investigation is necessary to understand the HIF-2α regulatory network.
The inhibitory effect of αTOA on HIF-2α can be realized via Phosphatidylinositol-3-Kinase and Protein Kinase B (Pi3K/Akt) signaling [
22], a key mechanism related to the proliferative fate of cells. Enhanced Pi3k/Akt pathway activation results in HIF-2α overexpression [
23], so one might suppose that the opposite action also operates, which is a hypothesis that remains to be explored. In addition, our proteomic analysis showed a downregulation of the Pi3K signaling pathway protein in CB CD34
+ cells after 3 days of culture with αTOA (data not shown).
Moreover, the inhibition of cyclin D1 by αTOA in CB CD34
+ cells could also be related to the Pi3K/Akt pathway [
24]. It has already been shown that γ-tocopherol inhibits cell cycle progression and cyclin D1 expression in human cancer cells [
25]. In our study, we show that αTOA can also modulate the cell cycle by decreasing cyclin D1 expression, most likely via the downregulation of the Pi3k/Akt pathway, which might explain the final effect of αTOA on the G0 accumulation of CD34
+ cells that we detected.
As a matter of fact, the culture that was not supplemented with αTOA should be considered rather as a vitamin E deficiency model [
3]. Indeed, our proteomic analysis (data not shown) confirms a high activity of adipocyte plasma membrane associated protein (APMAP), exhibiting a high arylesterase activity, as well as seven enzymes of the mevalonate pathway (whose precursor is acetate). Thus, as in other cell types [
2,
26], it is apparent that in CD34
+ cells, αTOA is hydrolyzed. In this view, αTOA supplementation yields αTOC, thus resolving αTOC deficiency and enabling the creation of a biomimetic environment as a better analogue of the physiological one. In this respect, one can hypothesize that one of the physiological roles of αTOA is related to HSC maintenance. The in vivo data presented here seem to be in favor of this hypothesis, since we found the signs of partial exhaustion of the primitive HSCs (in vivo repopulating cells), both in terms of their functional activity (in vivo repopulation test) as well as in terms of inflation of the phenotypically-defined bone marrow SLAM and SLK populations, which was not accompanied by an increase in engraftment capacity. One may further hypothesize that αTOC might protect the HSC from cycle-specific agents in vivo, or even from ionizing radiation, which remains to be explored. The enhanced compartment of SLAM and LSK (
Figure 7D,E) can be explained by the well-known phenomenon of phenotype/function dissociation [
27,
28]. Furthermore, we found an enhanced proliferative activity at the level of the committed progenitors in mice with αTOC deficiency (which is opposite to the ex vivo inhibition of proliferation by αTOA). Therefore, without αTOC, HSC and HPC accelerate their proliferation and lose their proliferative capacity. It should be stressed that in these experiments, we provoked only a relative deficiency (
Figure S4) of αTOC. Thus, the effects demonstrated here would most likely be even more pronounced in a real model of αTOC deficiency. If we consider these results bearing in mind the historical data concerning the clinical cases of αTOC deficiency (both in human and veterinary medicine), we can recognize an excessive proliferation of committed progenitors and precursor cells in view of an erythroid hyperplasia with multinucleated erythroid precursors in rhesus monkeys [
29] and rabbits [
30]. Other reports consider the hemolytic anemias in preterm human neonates [
31,
32] and in owl monkeys [
33]. Another interesting detail that we found is the phenomenon of the heart-shaped BFU-E colonies grown from the cells that were cultured in the presence of αTOA (
Figure S5). This phenomenon might reveal an effect on the asymmetrical proliferation of descending populations (CFU-E and precursors), or an influence of αTOA on the migration of progenitors or/and precursors, which all remains to be explored in more detail.
The improved maintenance of HSC was seen concerning the effects of αTOA on MSCs [
3] whose mitochondrial respiration was impacted, similarly to the results of our study. However, all other effects were different in comparison to those evidenced in CD34
+ cells, i.e., αTOA did not impact MSC cycling and did not have an effect on HIF-2, but it did stabilize HIF-1α, which seems to be related to the ROS increase detected at 24 h after αTOA treatment. Thus, the biochemical and molecular response of αTOA appears to be dependent on the cell type. However, similarities in the results for MSC and HSC seem to indicate a positive effect of αTOA on the maintenance of stem cells.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






