
Nanobodies for Medical Imaging: About Ready for Prime Time?

 ,
,
Abstract
:1. Introduction to Molecular Imaging
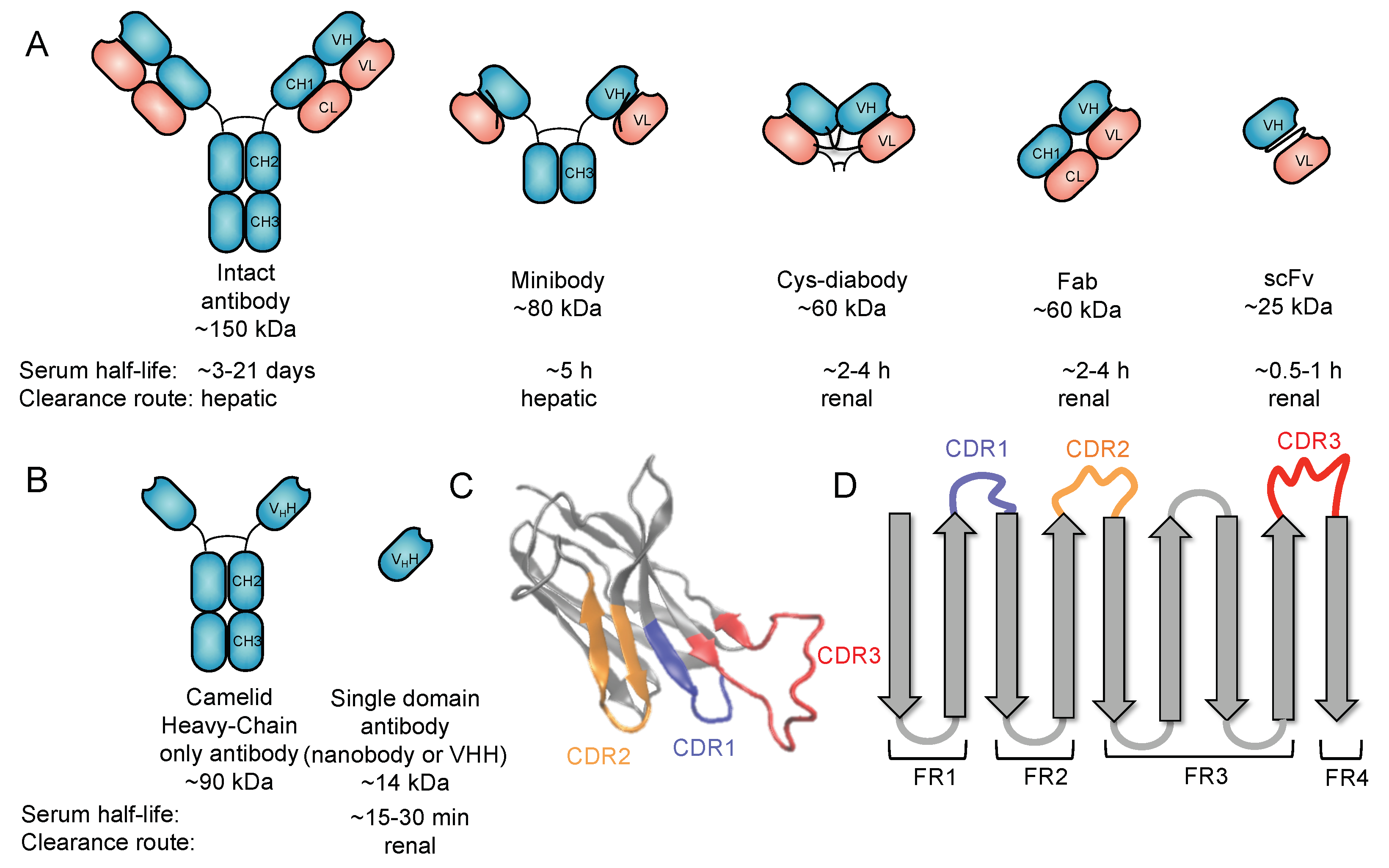
2. Use of Antibodies for Targeted Imaging
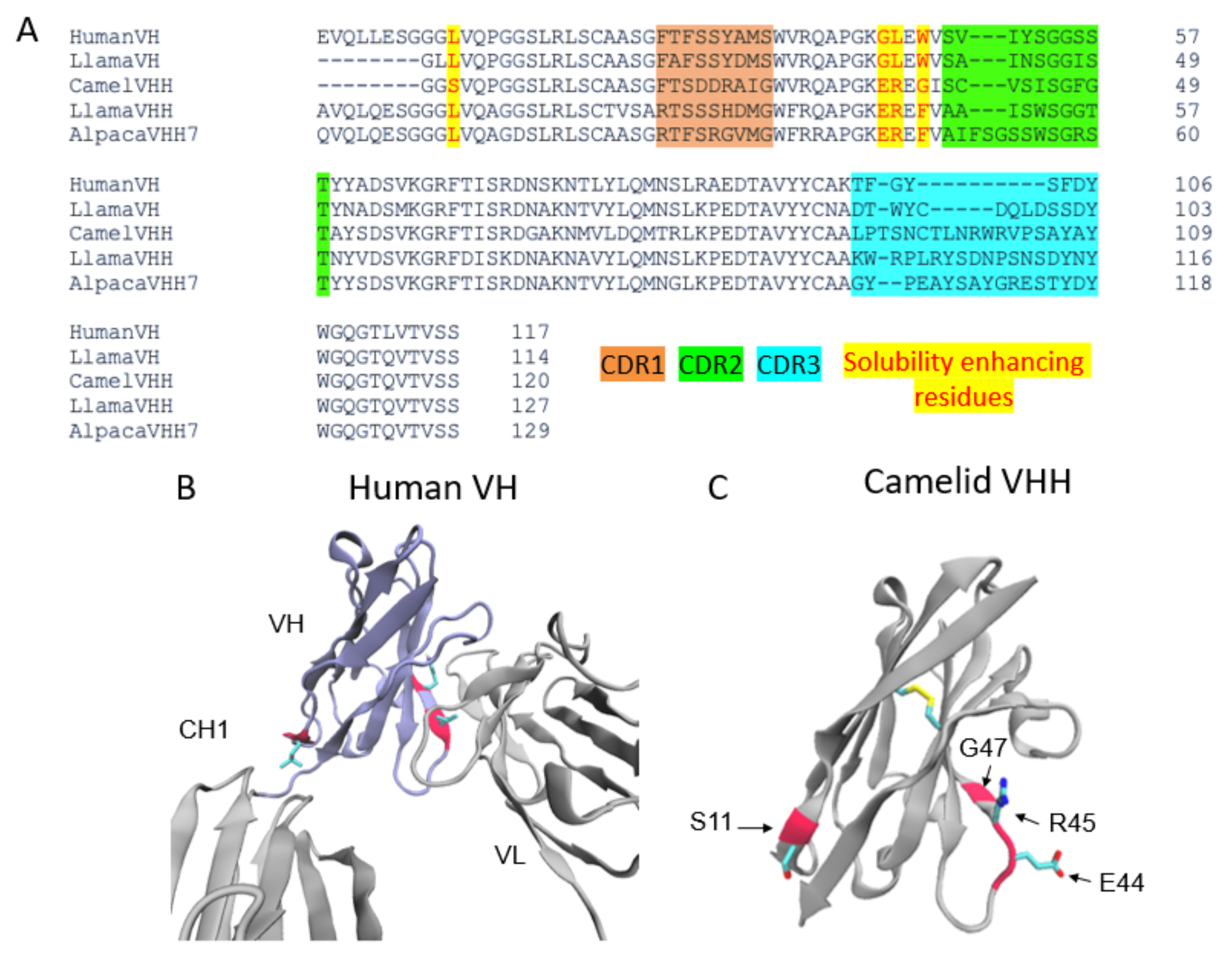
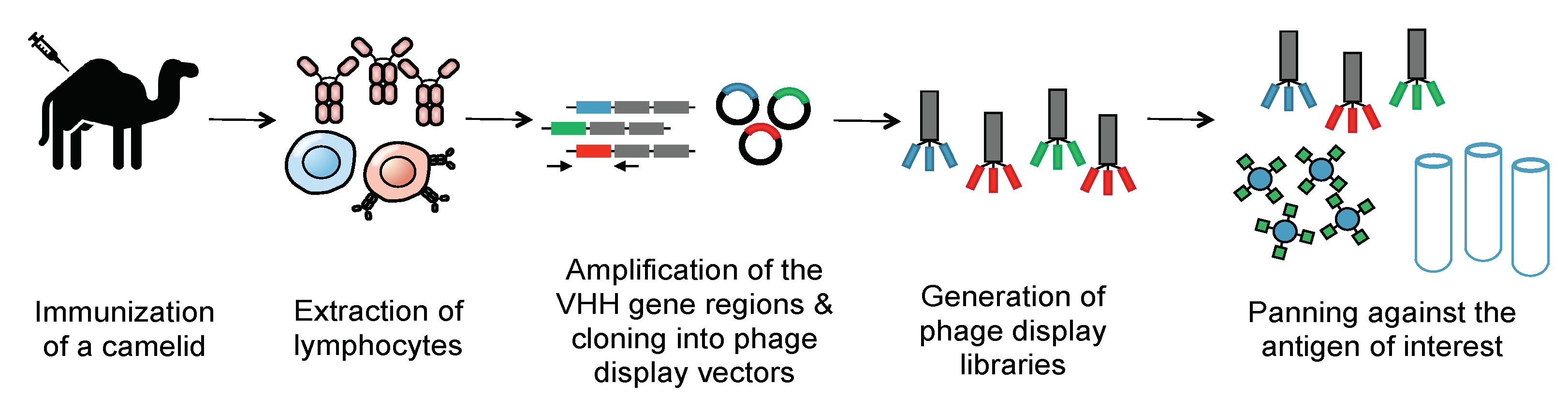
3. Nanobodies
4. Nanobodies as a Tool for Imaging Cancer
4.1. Imaging Cancer Cell Markers
4.1.1. EGFR
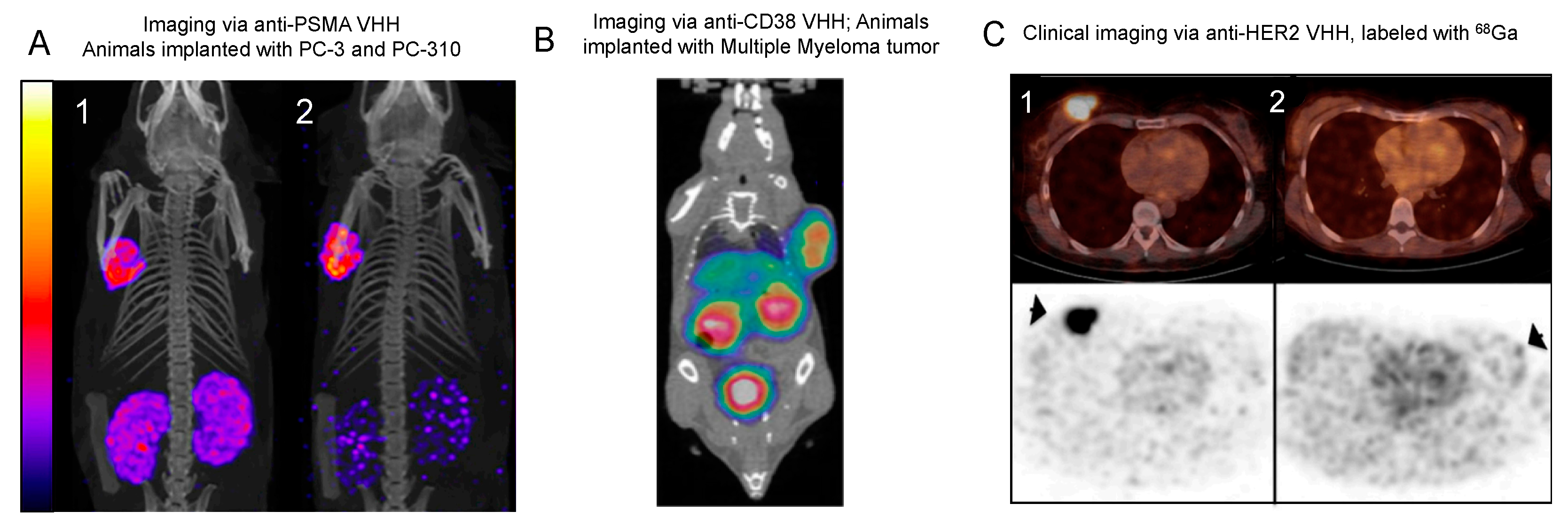
4.1.2. HER2
4.1.3. HER3
4.1.4. CEA
4.1.5. PSMA
4.1.6. HGF
4.1.7. CD20
4.1.8. CD38
4.1.9. Mesothelin
4.1.10. ECM Biomarkers: Imaging Cancer
4.2. Imaging Immune Checkpoint Markers
4.2.1. Background
4.2.2. PD-L1
4.2.3. CTLA-4
4.2.4. LAG-3
4.3. Imaging Immune Markers
4.3.1. Background
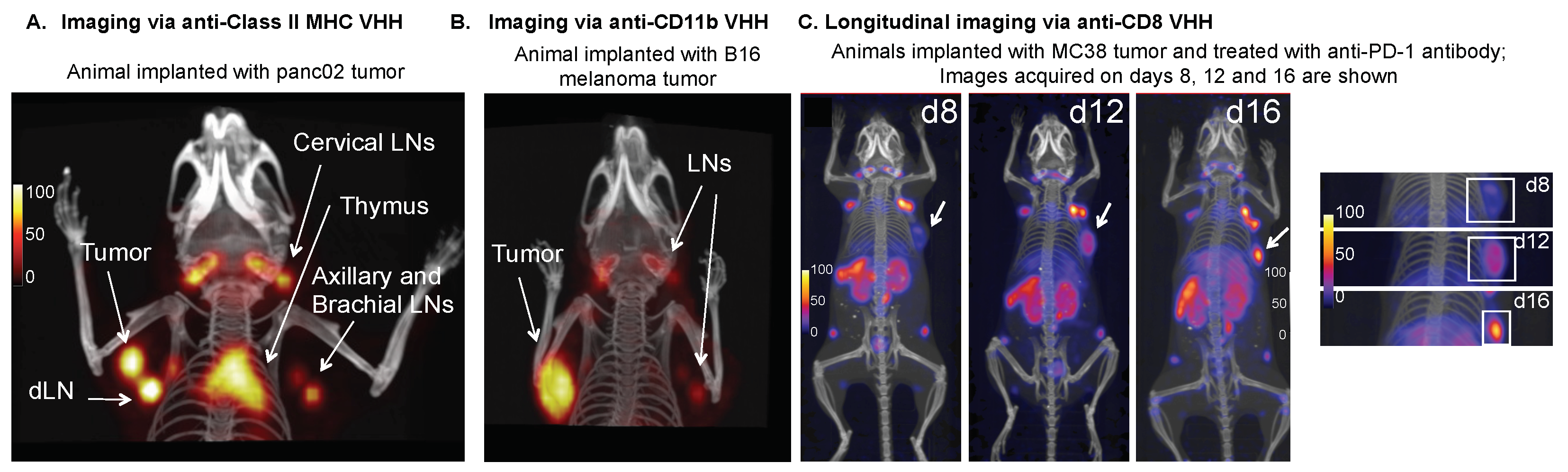
4.3.2. CD8
4.3.3. MHC Class II
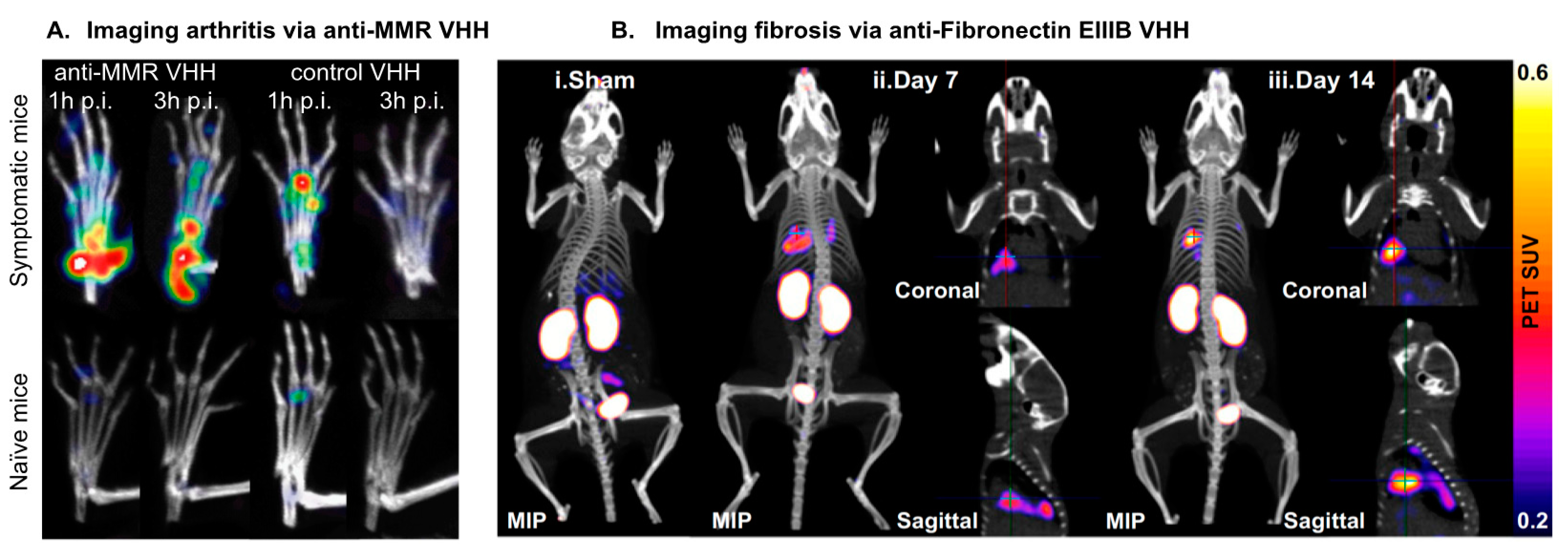
4.3.4. MMR
4.3.5. CD11b
4.4. Summary of Imaging Non-Cancer Targets
5. Nanobodies as a Tool for Imaging Non-Malignant Disease
5.1. ECM Biomarkers: Imaging Fibroses
5.2. VCAM-1: Imaging Cardiovascular Complications
5.3. MMR: Imaging Arthritis
5.4. αSyn: Tracking Neurodegenerative Disorders
5.5. DPP6: Imaging Insulin-Secreting Cells in Diabetes
5.6. KC: Imaging Liver Inflammation and Pathogenesis
5.7. Summary of Imaging Non-Cancer Targets
6. Conclusions and Future Directions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Agent | Reactivity | Clinical Trials: Stage and Status (If Applicable) | References |
|---|---|---|---|---|
| EGFR | 99mTc-8B6 | Human | Preclinical | [38] |
| 99mTc-7C12 | Human | Preclinical | [48] | |
| HER2 | 177Lu-2Rs15dHIS | Human | Preclinical | [55] |
| 18F-FB-2Rs15d | Murine | Preclinical | [56] | |
| 18F-RL-I-5F7 | Murine | Preclinical | [57] | |
| 68Ga-2Rs15d | Human | Clinical | [56,165] | |
| HER3 | 89Zr-MSB0010853 | Murine | Preclinical | [62] |
| CEA | 99mTc-NbCEA5 | Human | Preclinical | [65] |
| PSMA | 111In-JVZ007 | Human | Preclinical | [59] |
| HGF | 89Zr-1E2, 89Zr-6E10 | Human | Preclinical | [67] |
| CD20 | 68Ga-9079 | Human | Preclinical | [70] |
| CD38 | 68Ga-NOTA-Nb1053 | Murine | Preclinical | [60] |
| Mesothelin | 99mTc-A1, 99mTc-C6 | Human | Preclinical | [76] |
| MMR | 99mTc-d a-MMR Nb cl1 | Murine | Preclinical | [128,166] |
| 18 F-FB-anti-MMR 3.49 | Human, Murine | Preclinical | [123] | |
| 68Ga-NOTA-Anti-MMR-VHH2 | Human | Clinical, NCT04168528 (Active) | [124] | |
| MHC II | [18F]FDG -VHH7 | Murine | Preclinical | [117] |
| 64Cu- VHH4 | Human | Preclinical | [121] | |
| CD11b | 89Zr-VHHDC13 (PEGylated) | Murine | Preclinical | [115] |
| 18F-VHHDC13 | Human | Preclinical | [118] | |
| CD8 | 89Zr-VHH-X118 (PEGylated) | Murine | Preclinical | [43] |
| 68Ga-NOTA-SNA006 | Human | Preclinical | [119] | |
| Mouse Dendritic Cells | 99mTc-Nb-DC2.1 | Murine | Preclinical | [167] |
| 99mTc-Nb-DC1.8 | Murine | Preclinical | [167] | |
| PD-L1 | 18F-B3, 18F-A12, 64Cu-B3 | Murine | Preclinical | [95] |
| 99mTc-C3, 99mTc-C7, 99mTc-E2, 99mTc-E4, 99mTc-K2 | Murine | Preclinical | [92,93,94,168] | |
| 68Ga-NOTA-Nb109 | Human | Preclinical | [169] | |
| 99mTc-NM-01 | Human | Clinical, NCT02978196 (Concluded) | [98] | |
| 89Zr-envafolimab (Fc fusion) | Human | Clinical, NCT03638804 (Active) | [99,100] | |
| CTLA-4 | 18F-H11, 89Zr-H11 | Murine | Preclinical | [100,106] |
| LAG-3 | 99mTc-anti-moLAG-3 3206, 99mTc-anti-moLAG-3 3208, 99mTc-anti-moLAG-3 3132, 99mTc-anti-moLAG-3 3141 | Murine | Preclinical | [110,111] |
| VCAM-1 | 99m Tc-cAbVCAM1-5 | Human, Murine | Preclinical | [128,130,170,171] |
| FN-EIIIB (ECM) | 64 Cu-NJB2 | Human, Murine | Preclinical | [80] |
| αSyn | NbSyn2, NbSyn87 (fused to fluorescent proteins for imaging) | Human | Preclinical | [140,141] |
| DPP6 | 99m Tc-4hD29 | Human | Preclinical | [145] |
| Vsig4 | 99m Tc-NbV4 | Murine | Preclinical | [151,154] |
| Clec4F (KC) | 99m Tc-NbC4 | Murine | Preclinical | [151] |
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, L.H.; Seymour, L.; Litière, S.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1—Standardisation and Disease-Specific Adaptations: Perspectives from the RECIST Working Group. Eur. J. Cancer Oxf. Engl. 1990 2016, 62, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankoff, D.A.; Farwell, M.D.; Clark, A.S.; Pryma, D.A. Making Molecular Imaging a Clinical Tool for Precision Oncology: A Review. JAMA Oncol. 2017, 3, 695–701. [Google Scholar] [CrossRef]
- Aloya, R.; Shirvan, A.; Grimberg, H.; Reshef, A.; Levin, G.; Kidron, D.; Cohen, A.; Ziv, I. Molecular Imaging of Cell Death in Vivo by a Novel Small Molecule Probe. Apoptosis Int. J. Program. Cell Death 2006, 11, 2089–2101. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Xie, J.; Chen, X. Peptide-Based Probes for Targeted Molecular Imaging. Biochemistry 2010, 49, 1364–1376. [Google Scholar] [CrossRef] [Green Version]
- Chen, X. Protein and Peptide Probes for Molecular Imaging. Amino Acids 2011, 41, 1009–1012. [Google Scholar] [CrossRef] [Green Version]
- Thorek, D.L.J.; Chen, A.K.; Czupryna, J.; Tsourkas, A. Superparamagnetic Iron Oxide Nanoparticle Probes for Molecular Imaging. Ann. Biomed. Eng. 2006, 34, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.-R.; Byun, B.-H.; Hong, S.-P.; Chong, A.; Kim, J.; Yoo, S.-W.; Kang, S.-R.; Kim, D.-Y.; Song, H.-C.; Bom, H.-S.; et al. Comparison of 131I Whole-Body Imaging, 131I SPECT/CT, and 18F-FDG PET/CT in the Detection of Metastatic Thyroid Cancer. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Hartshorne, M.F. Single Photon Emission Computed Tomography. In Functional Brain Imaging; Elsevier: Amsterdam, The Netherlands, 1995; pp. 213–238. ISBN 978-0-8151-6509-5. [Google Scholar]
- Alauddin, M.M. Positron Emission Tomography (PET) Imaging with (18)F-Based Radiotracers. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 55–76. [Google Scholar] [PubMed]
- Bockisch, A.; Freudenberg, L.S.; Schmidt, D.; Kuwert, T. Hybrid Imaging by SPECT/CT and PET/CT: Proven Outcomes in Cancer Imaging. Semin. Nucl. Med. 2009, 39, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Van Valkenburgh, J.; Hong, X.; Conti, P.S.; Zhang, X.; Chen, K. Small Molecules as Theranostic Agents in Cancer Immunology. Theranostics 2019, 9, 7849–7871. [Google Scholar] [CrossRef]
- Vāvere, A.L.; Scott, P.J.H. Clinical Applications of Small-Molecule PET Radiotracers: Current Progress and Future Outlook. Semin. Nucl. Med. 2017, 47, 429–453. [Google Scholar] [CrossRef] [PubMed]
- Abousaway, O.; Rakhshandehroo, T.; Van den Abbeele, A.D.; Kircher, M.F.; Rashidian, M. Noninvasive Imaging of Cancer Immunotherapy. Nanotheranostics 2021, 5, 90–112. [Google Scholar] [CrossRef]
- Iravani, A.; Hicks, R.J. Imaging the Cancer Immune Environment and Its Response to Pharmacologic Intervention, Part 1: The Role of 18F-FDG PET/CT. J. Nucl. Med. 2020, 61, 943–950. [Google Scholar] [CrossRef]
- van Waarde, A.; Cobben, D.C.P.; Suurmeijer, A.J.H.; Maas, B.; Vaalburg, W.; de Vries, E.F.J.; Jager, P.L.; Hoekstra, H.J.; Elsinga, P.H. Selectivity of 18F-FLT and 18F-FDG for Differentiating Tumor from Inflammation in a Rodent Model. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2004, 45, 695–700. [Google Scholar]
- Khandani, A.H.; Dunphy, C.H.; Meteesatien, P.; Dufault, D.L.; Ivanovic, M.; Shea, T.C. Glut1 and Glut3 Expression in Lymphoma and Their Association with Tumor Intensity on 18F-Fluorodeoxyglucose Positron Emission Tomography. Nucl. Med. Commun. 2009, 30, 594–601. [Google Scholar] [CrossRef]
- Kaira, K.; Serizawa, M.; Koh, Y.; Takahashi, T.; Hanaoka, H.; Oriuchi, N.; Endo, M.; Kondo, H.; Nakajima, T.; Yamamoto, N. Relationship between 18F-FDG Uptake on Positron Emission Tomography and Molecular Biology in Malignant Pleural Mesothelioma. Eur. J. Cancer 2012, 48, 1244–1254. [Google Scholar] [CrossRef]
- Kawada, K.; Iwamoto, M.; Sakai, Y. Mechanisms Underlying 18F-Fluorodeoxyglucose Accumulation in Colorectal Cancer. World J. Radiol. 2016, 8, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, S.; Heinzmann, K.; Bollineni, V.R.; Kramer, G.M.; Liu, Y.; Waterton, J.C.; Aboagye, E.O.; Shields, A.F.; Soloviev, D.; Jacobs, A.H. Preclinical Applications of 3′-Deoxy-3′-[18F]Fluorothymidine in Oncology—A Systematic Review. Theranostics 2017, 7, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Barthel, H.; Cleij, M.C.; Collingridge, D.R.; Hutchinson, O.C.; Osman, S.; He, Q.; Luthra, S.K.; Brady, F.; Price, P.M.; Aboagye, E.O. 3′-Deoxy-3′-[18F]Fluorothymidine as a New Marker for Monitoring Tumor Response to Antiproliferative Therapy in Vivo with Positron Emission Tomography. Cancer Res. 2003, 63, 3791–3798. [Google Scholar] [PubMed]
- Rashidian, M.; Ploegh, H. Nanobodies as Non-Invasive Imaging Tools. Immuno-Oncol. Technol. 2020, 7, 2–14. [Google Scholar] [CrossRef]
- Campos, C.D.M.; Jackson, J.M.; Witek, M.A.; Soper, S.A. Molecular Profiling of Liquid Biopsy Samples for Precision Medicine. Cancer J. Sudbury Mass 2018, 24, 93–103. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic Heterogeneity of Breast Cancer: Molecular Mechanism and Potential Therapeutic Targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor Heterogeneity: Causes and Consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Jayson, G.C.; Zweit, J.; Jackson, A.; Mulatero, C.; Julyan, P.; Ranson, M.; Broughton, L.; Wagstaff, J.; Hakannson, L.; Groenewegen, G.; et al. Molecular Imaging and Biological Evaluation of HuMV833 Anti-VEGF Antibody: Implications for Trial Design of Antiangiogenic Antibodies. J. Natl. Cancer Inst. 2002, 94, 1484–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody Tumor Penetration: Transport Opposed by Systemic and Antigen-Mediated Clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.-Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Lipovsek, D. Adnectins: Engineered Target-Binding Protein Therapeutics. Protein Eng. Des. Sel. PEDS 2011, 24, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, D.J.; Smith, R.A.; Morin, P.; Lipovšek, D.; Gokemeijer, J.; Cohen, D.; Lafont, V.; Tran, T.; Cole, E.L.; Wright, M.; et al. Synthesis and Biologic Evaluation of a Novel 18 F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J. Nucl. Med. 2018, 59, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A New Generation of Protein Therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.; Renko, M.; Završnik, J.; Turk, D.; Seeger, M.A.; Vasiljeva, O.; Grütter, M.G.; Turk, V.; Turk, B. Non-Invasive in Vivo Imaging of Tumour-Associated Cathepsin B by a Highly Selective Inhibitory DARPin. Theranostics 2017, 7, 2806–2821. [Google Scholar] [CrossRef] [Green Version]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.F.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A General Protocol for the Generation of Nanobodies for Structural Biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef]
- Debie, P.; Devoogdt, N.; Hernot, S. Targeted Nanobody-Based Molecular Tracers for Nuclear Imaging and Image-Guided Surgery. Antibodies 2019, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, L.S.; Colwell, L.J. Comparative Analysis of Nanobody Sequence and Structure Data. Proteins Struct. Funct. Bioinform. 2018, 86, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. A Guide to: Generation and Design of Nanobodies. FEBS J. 2020. [Google Scholar] [CrossRef]
- Fleetwood, F.; Devoogdt, N.; Pellis, M.; Wernery, U.; Muyldermans, S.; Ståhl, S.; Löfblom, J. Surface Display of a Single-Domain Antibody Library on Gram-Positive Bacteria. Cell. Mol. Life Sci. CMLS 2013, 70, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Koide, S. Affinity Maturation of Single-Domain Antibodies by Yeast Surface Display. Methods Mol. Biol. Clifton NJ 2012, 911, 431–443. [Google Scholar] [CrossRef]
- Salema, V.; Fernández, L.Á. Escherichia Coli Surface Display for the Selection of Nanobodies. Microb. Biotechnol. 2017, 10, 1468–1484. [Google Scholar] [CrossRef]
- Salema, V.; Mañas, C.; Cerdán, L.; Piñero-Lambea, C.; Marín, E.; Roovers, R.C.; Van Bergen En Henegouwen, P.M.P.; Fernández, L.Á. High Affinity Nanobodies against Human Epidermal Growth Factor Receptor Selected on Cells by E. coli Display. mAbs 2016, 8, 1286–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast Surface Display Platform for Rapid Discovery of Conformationally Selective Nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Higashikawa, K.; Yagi, K.; Watanabe, K.; Kamino, S.; Ueda, M.; Hiromura, M.; Enomoto, S. 64Cu-DOTA-Anti-CTLA-4 MAb Enabled PET Visualization of CTLA-4 on the T-Cell Infiltrating Tumor Tissues. PLoS ONE 2014, 9, e109866. [Google Scholar] [CrossRef]
- Rashidian, M.; Ingram, J.R.; Dougan, M.; Dongre, A.; Whang, K.A.; LeGall, C.; Cragnolini, J.J.; Bierie, B.; Gostissa, M.; Gorman, J.; et al. Predicting the Response to CTLA-4 Blockade by Longitudinal Noninvasive Monitoring of CD8 T Cells. J. Exp. Med. 2017, 214, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; Devoogdt, N.; Hernot, S.; Vaneycken, I.; D’Huyvetter, M.; De Vos, J.; Massa, S.; Lahoutte, T.; Caveliers, V. Site-Specific Labeling of His-Tagged Nanobodies with 99mTc: A Practical Guide. Methods Mol. Biol. Clifton NJ 2012, 911, 485–490. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Selvaggi, G.; Novello, S.; Hirsch, F.R. The Biology of Epidermal Growth Factor Receptor in Lung Cancer. Clin. Cancer Res. 2004, 10, 4227s–4232s. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.A.; Hussain, Z.F.; Aijaz, S.; Irfan, M.; Khan, E.Y.; Naz, S.; Faridi, N.; Khan, A.; Edhi, M.M. Immunohistochemical Expression of Epidermal Growth Factor Receptor (EGFR) in South Asian Head and Neck Squamous Cell Carcinoma: Association with Various Risk Factors and Clinico-Pathologic and Prognostic Parameters. World J. Surg. Oncol. 2018, 16, 118. [Google Scholar] [CrossRef]
- Huang, L.; Gainkam, L.O.T.; Caveliers, V.; Vanhove, C.; Keyaerts, M.; De Baetselier, P.; Bossuyt, A.; Revets, H.; Lahoutte, T. SPECT Imaging with 99mTc-Labeled EGFR-Specific Nanobody for In Vivo Monitoring of EGFR Expression. Mol. Imaging Biol. 2008, 10, 167–175. [Google Scholar] [CrossRef]
- Gainkam, L.O.T.; Keyaerts, M.; Caveliers, V.; Devoogdt, N.; Vanhove, C.; Van Grunsven, L.; Muyldermans, S.; Lahoutte, T. Correlation between Epidermal Growth Factor Receptor-Specific Nanobody Uptake and Tumor Burden: A Tool for Noninvasive Monitoring of Tumor Response to Therapy. Mol. Imaging Biol. 2011, 13, 940–948. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast Cancer Subtypes Based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Asif, H.M.; Sultana, S.; Ahmed, S.; Akhtar, N.; Tariq, M. HER-2 Positive Breast Cancer—A Mini-Review. Asian Pac. J. Cancer Prev. 2016, 17, 1609–1615. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-Y.; Bang, Y.-J. HER2-Targeted Therapies—A Role beyond Breast Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the Treatment of Breast Cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef]
- Albanell, J.; Codony, J.; Rovira, A.; Mellado, B.; Gascón, P. Mechanism of Action of Anti-HER2 Monoclonal Antibodies: Scientific Update on Trastuzumab and 2C4. Adv. Exp. Med. Biol. 2003, 532, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Hyman, D.M.; Ross, D.S.; Corben, A.; Chandarlapaty, S.; Goldfarb, S.; McArthur, H.; Erinjeri, J.P.; Solomon, S.B.; Kolb, H.; et al. Detection of HER2-Positive Metastases in Patients with HER2-Negative Primary Breast Cancer Using 89Zr-Trastuzumab PET/CT. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 1523–1528. [Google Scholar] [CrossRef] [Green Version]
- D’Huyvetter, M.; Aerts, A.; Xavier, C.; Vaneycken, I.; Devoogdt, N.; Gijs, M.; Impens, N.; Baatout, S.; Ponsard, B.; Muyldermans, S.; et al. Development of 177Lu-Nanobodies for Radioimmunotherapy of HER2-Positive Breast Cancer: Evaluation of Different Bifunctional Chelators: 177LU-NANOBODIES FOR RADIOIMMUNOTHERAPY. Contrast Media Mol. Imaging 2012, 7, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; Blykers, A.; Vaneycken, I.; D’Huyvetter, M.; Heemskerk, J.; Lahoutte, T.; Devoogdt, N.; Caveliers, V. (18)F-Nanobody for PET Imaging of HER2 Overexpressing Tumors. Nucl. Med. Biol. 2016, 43, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, G.; McDougald, D.; Choi, J.; Koumarianou, E.; Weitzel, D.; Osada, T.; Lyerly, H.K.; Zalutsky, M.R. Preclinical Evaluation of 18F-Labeled Anti-HER2 Nanobody Conjugates for Imaging HER2 Receptor Expression by Immuno-PET. J. Nucl. Med. 2016, 57, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Chatalic, K.L.S.; Veldhoven-Zweistra, J.; Bolkestein, M.; Hoeben, S.; Koning, G.A.; Boerman, O.C.; de Jong, M.; van Weerden, W.M. A Novel 111In-Labeled Anti-Prostate-Specific Membrane Antigen Nanobody for Targeted SPECT/CT Imaging of Prostate Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1094–1099. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, Y.; Hou, Y.N.; Liu, Q.; Zhang, D.; Zhao, H.; Zhang, Y.; An, S.; Li, L.; Hou, J.; et al. ImmunoPET Imaging of Multiple Myeloma with [68Ga]Ga-NOTA-Nb1053. Eur. J. Nucl. Med. Mol. Imaging 2021. [CrossRef]
- Gaborit, N.; Abdul-Hai, A.; Mancini, M.; Lindzen, M.; Lavi, S.; Leitner, O.; Mounier, L.; Chentouf, M.; Dunoyer, S.; Ghosh, M.; et al. Examination of HER3 Targeting in Cancer Using Monoclonal Antibodies. Proc. Natl. Acad. Sci. USA 2015, 112, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Warnders, F.J.; Terwisscha van Scheltinga, A.G.T.; Knuehl, C.; van Roy, M.; de Vries, E.F.J.; Kosterink, J.G.W.; de Vries, E.G.E.; Lub-de Hooge, M.N. Human Epidermal Growth Factor Receptor 3-Specific Tumor Uptake and Biodistribution of 89Zr-MSB0010853 Visualized by Real-Time and Noninvasive PET Imaging. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1210–1215. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, L.H.; Ribas, A.; Economou, J.S. DNA and Dendritic Cell-Based Genetic Immunization Against Cancer. In Gene Therapy of Cancer; Elsevier: Amsterdam, The Netherlands, 2002; pp. 179–198. ISBN 978-0-12-437551-2. [Google Scholar]
- Kokkonen, N.; Ulibarri, I.F.; Kauppila, A.; Luosujärvi, H.; Rivinoja, A.; Pospiech, H.; Kellokumpu, I.; Kellokumpu, S. Hypoxia Upregulates Carcinoembryonic Antigen Expression in Cancer Cells. Int. J. Cancer 2007, 121, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Vaneycken, I.; Govaert, J.; Vincke, C.; Caveliers, V.; Lahoutte, T.; De Baetselier, P.; Raes, G.; Bossuyt, A.; Muyldermans, S.; Devoogdt, N. In Vitro Analysis and in Vivo Tumor Targeting of a Humanized, Grafted Nanobody in Mice Using Pinhole SPECT/Micro-CT. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorff, T.B.; Fanti, S.; Farolfi, A.; Reiter, R.E.; Sadun, T.Y.; Sartor, O. The Evolving Role of Prostate-Specific Membrane Antigen–Based Diagnostics and Therapeutics in Prostate Cancer. Am. Soc. Clin. Oncol. Educ. Book 2019, 321–330. [Google Scholar] [CrossRef]
- Vosjan, M.J.W.D.; Vercammen, J.; Kolkman, J.A.; Stigter-van Walsum, M.; Revets, H.; van Dongen, G.A.M.S. Nanobodies Targeting the Hepatocyte Growth Factor: Potential New Drugs for Molecular Cancer Therapy. Mol. Cancer Ther. 2012, 11, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Shanehbandi, D.; Majidi, J.; Kazemi, T.; Baradaran, B.; Aghebati-Maleki, L. CD20-Based Immunotherapy of B-Cell Derived Hematologic Malignancies. Curr. Cancer Drug Targets 2017, 17, 423–444. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasniqi, A.; D’Huyvetter, M.; Xavier, C.; Van der Jeught, K.; Muyldermans, S.; Van Der Heyden, J.; Lahoutte, T.; Tavernier, J.; Devoogdt, N. Theranostic Radiolabeled Anti-CD20 SdAb for Targeted Radionuclide Therapy of Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2017, 16, 2828–2839. [Google Scholar] [CrossRef] [Green Version]
- Karam, M.; Novak, L.; Cyriac, J.; Ali, A.; Nazeer, T.; Nugent, F. Role of Fluorine-18 Fluoro-Deoxyglucose Positron Emission Tomography Scan in the Evaluation and Follow-up of Patients with Low-Grade Lymphomas. Cancer 2006, 107, 175–183. [Google Scholar] [CrossRef]
- Costa, F.; Dalla Palma, B.; Giuliani, N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells 2019, 8, 1632. [Google Scholar] [CrossRef] [Green Version]
- Ostendorf, L.; Burns, M.; Durek, P.; Heinz, G.A.; Heinrich, F.; Garantziotis, P.; Enghard, P.; Richter, U.; Biesen, R.; Schneider, U.; et al. Targeting CD38 with Daratumumab in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 383, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Terpos, E.; Nanni, C.; Moreau, P.; Lentzsch, S.; Zweegman, S.; Hillengass, J.; Engelhardt, M.; Usmani, S.Z.; Vesole, D.H.; et al. Role of 18F-FDG PET/CT in the Diagnosis and Management of Multiple Myeloma and Other Plasma Cell Disorders: A Consensus Statement by the International Myeloma Working Group. Lancet Oncol. 2017, 18, e206–e217. [Google Scholar] [CrossRef]
- Hassan, R.; Ho, M. Mesothelin Targeted Cancer Immunotherapy. Eur. J. Cancer Oxf. Engl. 1990 2008, 44, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montemagno, C.; Bacot, S.; Ahmadi, M.; Kerfelec, B.; Baty, D.; Debiossat, M.; Soubies, A.; Perret, P.; Riou, L.; Fagret, D.; et al. Preclinical Evaluation of Mesothelin-Specific Ligands for SPECT Imaging of Triple-Negative Breast Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Teti, A. Regulation of Cellular Functions by Extracellular Matrix. J. Am. Soc. Nephrol. JASN 1992, 2, S83–S87. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jailkhani, N.; Ingram, J.R.; Rashidian, M.; Rickelt, S.; Tian, C.; Mak, H.; Jiang, Z.; Ploegh, H.L.; Hynes, R.O. Noninvasive Imaging of Tumor Progression, Metastasis, and Fibrosis Using a Nanobody Targeting the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2019, 116, 14181–14190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberstein, P.E.; Olive, K.P. Pancreatic Cancer: Why Is It so Hard to Treat? Ther. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [Green Version]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse Effects of Immune-Checkpoint Inhibitors: Epidemiology, Management and Surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity Associated with CTLA4 and PD1 Blocking Immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, M.; Sholl, L.M.; Hodi, F.S.; Hatabu, H.; Ramaiya, N.H. Anti-PD-1-Related Pneumonitis during Cancer Immunotherapy. N. Engl. J. Med. 2015, 373, 288–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 Expression in Human Cancers and Its Association with Clinical Outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Long-Mira, E.; Bence, C.; Butori, C.; Lassalle, S.; Bouhlel, L.; Fazzalari, L.; Zahaf, K.; Lalvée, S.; Washetine, K.; et al. Comparative Study of the PD-L1 Status between Surgically Resected Specimens and Matched Biopsies of NSCLC Patients Reveal Major Discordances: A Potential Issue for Anti-PD-L1 Therapeutic Strategies. Ann. Oncol. 2016, 27, 147–153. [Google Scholar] [CrossRef]
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Nguyen, T.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K.; Devoogdt, N. Non-Invasive Assessment of Murine PD-L1 Levels in Syngeneic Tumor Models by Nuclear Imaging with Nanobody Tracers. Oncotarget 2017, 8, 41932–41946. [Google Scholar] [CrossRef] [Green Version]
- Broos, K.; Lecocq, Q.; Raes, G.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Noninvasive Imaging of the PD-1:PD-L1 Immune Checkpoint: Embracing Nuclear Medicine for the Benefit of Personalized Immunotherapy. Theranostics 2018, 8, 3559–3570. [Google Scholar] [CrossRef] [PubMed]
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-Human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.R.; Dougan, M.; Rashidian, M.; Knoll, M.; Keliher, E.J.; Garrett, S.; Garforth, S.; Blomberg, O.S.; Espinosa, C.; Bhan, A.; et al. PD-L1 Is an Activation-Independent Marker of Brown Adipocytes. Nat. Commun. 2017, 8, 647. [Google Scholar] [CrossRef]
- Helou, D.G.; Shafiei-Jahani, P.; Lo, R.; Howard, E.; Hurrell, B.P.; Galle-Treger, L.; Painter, J.D.; Lewis, G.; Soroosh, P.; Sharpe, A.H.; et al. PD-1 Pathway Regulates ILC2 Metabolism and PD-1 Agonist Treatment Ameliorates Airway Hyperreactivity. Nat. Commun. 2020, 11, 3998. [Google Scholar] [CrossRef] [PubMed]
- Qorraj, M.; Bruns, H.; Böttcher, M.; Weigand, L.; Saul, D.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. The PD-1/PD-L1 Axis Contributes to Immune Metabolic Dysfunctions of Monocytes in Chronic Lymphocytic Leukemia. Leukemia 2017, 31, 470–478. [Google Scholar] [CrossRef]
- Xing, Y.; Chand, G.; Liu, C.; Cook, G.J.R.; O’Doherty, J.; Zhao, L.; Wong, N.C.L.; Meszaros, L.K.; Ting, H.H.; Zhao, J. Early Phase I Study of a 99mTc-Labeled Anti–Programmed Death Ligand-1 (PD-L1) Single-Domain Antibody in SPECT/CT Assessment of PD-L1 Expression in Non–Small Cell Lung Cancer. J. Nucl. Med. 2019, 60, 1213–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zou, S.; Cheng, S.; Song, S.; Wang, P.; Zhu, X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol. Pharm. 2019, 16, 3469–3476. [Google Scholar] [CrossRef]
- Li, D.; Cheng, S.; Zou, S.; Zhu, D.; Zhu, T.; Wang, P.; Zhu, X. Immuno-PET Imaging of 89Zr Labeled Anti-PD-L1 Domain Antibody. Mol. Pharm. 2018, 15, 1674–1681. [Google Scholar] [CrossRef]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-Atezolizumab Imaging as a Non-Invasive Approach to Assess Clinical Response to PD-L1 Blockade in Cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, E.-E.; Kilvaer, T.K.; Rakaee, M.; Richardsen, E.; Hald, S.M.; Andersen, S.; Busund, L.-T.; Bremnes, R.M.; Donnem, T. CTLA-4 Expression in the Non-Small Cell Lung Cancer Patient Tumor Microenvironment: Diverging Prognostic Impact in Primary Tumors and Lymph Node Metastases. Cancer Immunol. Immunother. 2017, 66, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hou, X.; Yang, X.; Liu, A.; Tang, Z.; Mo, F.; Yin, S.; Lu, X. Highly Sensitive Detection of CTLA-4-Positive T-Cell Subgroups Based on Nanobody and Fluorescent Carbon Quantum Dots. Oncol. Lett. 2019, 18, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Ingram, J.R.; Blomberg, O.S.; Rashidian, M.; Ali, L.; Garforth, S.; Fedorov, E.; Fedorov, A.A.; Bonanno, J.B.; Le Gall, C.; Crowley, S.; et al. Anti-CTLA-4 Therapy Requires an Fc Domain for Efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, 3912–3917. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.-P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 Defines Exhaustion of Intratumoral PD-1+ T Cells and Correlates with Poor Outcome in Follicular Lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef] [Green Version]
- Datar, I.; Sanmamed, M.F.; Wang, J.; Henick, B.S.; Choi, J.; Badri, T.; Dong, W.; Mani, N.; Toki, M.; Mejías, L.D.; et al. Expression Analysis and Significance of PD-1, LAG-3, and TIM-3 in Human Non-Small Cell Lung Cancer Using Spatially Resolved and Multiparametric Single-Cell Analysis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4663–4673. [Google Scholar] [CrossRef]
- Lecocq, Q.; Zeven, K.; De Vlaeminck, Y.; Martens, S.; Massa, S.; Goyvaerts, C.; Raes, G.; Keyaerts, M.; Breckpot, K.; Devoogdt, N. Noninvasive Imaging of the Immune Checkpoint LAG-3 Using Nanobodies, from Development to Pre-Clinical Use. Biomolecules 2019, 9, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecocq, Q.; Awad, R.M.; De Vlaeminck, Y.; De Mey, W.; Ertveldt, T.; Goyvaerts, C.; Raes, G.; Thielemans, K.; Keyaerts, M.; Devoogdt, N.; et al. Nanobody Nuclear Imaging Allows Noninvasive Quantification of LAG-3 Expression by Tumor-Infiltrating Leukocytes and Predicts Response of Immune Checkpoint Blockade. J. Nucl. Med. 2021. [Google Scholar] [CrossRef]
- Farwell, M.; Gamache, R.; Pandit-Taskar, N.; Postow, M.; Gordon, M.; Wilson, I.; Mascioni, A.; Wu, A.; Le, W.; Weiss, A.; et al. 294 CD8 PET Imaging of Tumor Infiltrating T Cells in Advanced Solid Tumors: A Phase I First-in-Human Study of 89Zr-IAB22M2C, a Radiolabeled Anti-CD8 Minibody. J. Immunother. Cancer 2020, 8, A321. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; Postow, M.; Hellmann, M.; Harding, J.; Barker, C.; O’Donoghue, J.; Ziolkowska, M.; Ruan, S.; Lyashchenko, S.; Tsai, F.; et al. First-in-Human Imaging with 89Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zito Marino, F.; Ascierto, P.A.; Rossi, G.; Staibano, S.; Montella, M.; Russo, D.; Alfano, R.; Morabito, A.; Botti, G.; Franco, R. Are Tumor-Infiltrating Lymphocytes Protagonists or Background Actors in Patient Selection for Cancer Immunotherapy? Expert Opin. Biol. Ther. 2017, 17, 735–746. [Google Scholar] [CrossRef]
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET Identifies the Myeloid Compartment as a Key Contributor to the Outcome of the Antitumor Response under PD-1 Blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971–16980. [Google Scholar] [CrossRef] [Green Version]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.; Edens, J.G.; Tas, J.M.G.; Victora, G.; et al. The Use of (18)F-2-Fluorodeoxyglucose (FDG) to Label Antibody Fragments for Immuno-PET of Pancreatic Cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Rashidian, M.; Keliher, E.J.; Bilate, A.M.; Duarte, J.N.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Cragnolini, J.; Swee, L.K.; Victora, G.D.; Weissleder, R.; et al. Noninvasive Imaging of Immune Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6146–6151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wang, C.; Yang, Y.; Sun, Y.; Wei, W.; Wang, C.; Wan, L.; Zhu, C.; Li, L.; Huang, G.; et al. ImmunoPET Imaging of Human CD8+ T Cells with Novel 68Ga-Labeled Nanobody Companion Diagnostic Agents. J. Nanobiotechnology 2021, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Krekorian, M.; Fruhwirth, G.O.; Srinivas, M.; Figdor, C.G.; Heskamp, S.; Witney, T.H.; Aarntzen, E.H.J.G. Imaging of T-Cells and Their Responses during Anti-Cancer Immunotherapy. Theranostics 2019, 9, 7924–7947. [Google Scholar] [CrossRef]
- Van Elssen, C.H.M.J.; Rashidian, M.; Vrbanac, V.; Wucherpfennig, K.W.; Habre, Z.E.; Sticht, J.; Freund, C.; Jacobsen, J.T.; Cragnolini, J.; Ingram, J.; et al. Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1003–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, J.L.M.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-Host Disease. Lancet Lond. Engl. 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Blykers, A.; Schoonooghe, S.; Xavier, C.; D’hoe, K.; Laoui, D.; D’Huyvetter, M.; Vaneycken, I.; Cleeren, F.; Bormans, G.; Heemskerk, J.; et al. PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1265–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.; Blykers, A.; Laoui, D.; Bolli, E.; Vaneyken, I.; Bridoux, J.; Baudhuin, H.; Raes, G.; Everaert, H.; Movahedi, K.; et al. Clinical Translation of [68Ga]Ga-NOTA-Anti-MMR-SdAb for PET/CT Imaging of Protumorigenic Macrophages. Mol. Imaging Biol. 2019, 21, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, H.M.; Carpenter, D.C.; Stahl, J.M.; Zhang, W.; Hynicka, W.P.; Griswold, D.E. Human Granulocyte CD11b Expression as a Pharmacodynamic Biomarker of Inflammation. J. Immunol. Methods 2000, 240, 125–132. [Google Scholar] [CrossRef]
- Sharma, M.; Tiwari, A.; Sharma, S.; Bhoria, P.; Gupta, V.; Gupta, A.; Luthra-Guptasarma, M. Fibrotic Remodeling of the Extracellular Matrix through a Novel (Engineered, Dual-Function) Antibody Reactive to a Cryptic Epitope on the N-Terminal 30 KDa Fragment of Fibronectin. PLoS ONE 2013, 8, e69343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wight, T.N.; Potter-Perigo, S. The Extracellular Matrix: An Active or Passive Player in Fibrosis? Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G950–G955. [Google Scholar] [CrossRef] [Green Version]
- Put, S.; Schoonooghe, S.; Devoogdt, N.; Schurgers, E.; Avau, A.; Mitera, T.; D’Huyvetter, M.; De Baetselier, P.; Raes, G.; Lahoutte, T.; et al. SPECT Imaging of Joint Inflammation with Nanobodies Targeting the Macrophage Mannose Receptor in a Mouse Model for Rheumatoid Arthritis. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2013, 54, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [Green Version]
- Broisat, A.; Hernot, S.; Toczek, J.; De Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies Targeting Mouse/Human VCAM1 for the Nuclear Imaging of Atherosclerotic Lesions. Circ. Res. 2012, 110, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Punjabi, M.; Xu, L.; Ochoa-Espinosa, A.; Kosareva, A.; Wolff, T.; Murtaja, A.; Broisat, A.; Devoogdt, N.; Kaufmann, B.A. Ultrasound Molecular Imaging of Atherosclerosis With Nanobodies: Translatable Microbubble Targeting Murine and Human VCAM (Vascular Cell Adhesion Molecule) 1. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2520–2530. [Google Scholar] [CrossRef]
- Iiyama, K.; Hajra, L.; Iiyama, M.; Li, H.; DiChiara, M.; Medoff, B.D.; Cybulsky, M.I. Patterns of Vascular Cell Adhesion Molecule-1 and Intercellular Adhesion Molecule-1 Expression in Rabbit and Mouse Atherosclerotic Lesions and at Sites Predisposed to Lesion Formation. Circ. Res. 1999, 85, 199–207. [Google Scholar] [CrossRef]
- Huo, Y.; Hafezi-Moghadam, A.; Ley, K. Role of Vascular Cell Adhesion Molecule-1 and Fibronectin Connecting Segment-1 in Monocyte Rolling and Adhesion on Early Atherosclerotic Lesions. Circ. Res. 2000, 87, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, B.A.; Carr, C.L.; Belcik, J.T.; Xie, A.; Yue, Q.; Chadderdon, S.; Caplan, E.S.; Khangura, J.; Bullens, S.; Bunting, S.; et al. Molecular Imaging of the Initial Inflammatory Response in Atherosclerosis: Implications for Early Detection of Disease. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic Antibodies: Successes, Limitations and Hopes for the Future: Therapeutic Antibodies: An Update. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Kaul, S. Myocardial Contrast Echocardiography: A 25-Year Retrospective. Circulation 2008, 118, 291–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid Arthritis: Pathological Mechanisms and Modern Pharmacologic Therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Put, S.; Westhovens, R.; Lahoutte, T.; Matthys, P. Molecular Imaging of Rheumatoid Arthritis: Emerging Markers, Tools, and Techniques. Arthritis Res. Ther. 2014, 16, 208. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of Alpha-Synuclein in Lewy Bodies of Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Gerdes, C.; Waal, N.; Offner, T.; Fornasiero, E.F.; Wender, N.; Verbarg, H.; Manzini, I.; Trenkwalder, C.; Mollenhauer, B.; Strohäker, T.; et al. A Nanobody-Based Fluorescent Reporter Reveals Human α-Synuclein in the Cell Cytosol. Nat. Commun. 2020, 11, 2729. [Google Scholar] [CrossRef] [PubMed]
- Iljina, M.; Hong, L.; Horrocks, M.H.; Ludtmann, M.H.; Choi, M.L.; Hughes, C.D.; Ruggeri, F.S.; Guilliams, T.; Buell, A.K.; Lee, J.-E.; et al. Nanobodies Raised against Monomeric Ɑ-Synuclein Inhibit Fibril Formation and Destabilize Toxic Oligomeric Species. BMC Biol. 2017, 15, 57. [Google Scholar] [CrossRef] [Green Version]
- Der-Sarkissian, A.; Jao, C.C.; Chen, J.; Langen, R. Structural Organization of Alpha-Synuclein Fibrils Studied by Site-Directed Spin Labeling. J. Biol. Chem. 2003, 278, 37530–37535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maass, F.; Schulz, I.; Lingor, P.; Mollenhauer, B.; Bähr, M. Cerebrospinal Fluid Biomarker for Parkinson’s Disease: An Overview. Mol. Cell. Neurosci. 2019, 97, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balhuizen, A.; Massa, S.; Mathijs, I.; Turatsinze, J.-V.; De Vos, J.; Demine, S.; Xavier, C.; Villate, O.; Millard, I.; Egrise, D.; et al. A Nanobody-Based Tracer Targeting DPP6 for Non-Invasive Imaging of Human Pancreatic Endocrine Cells. Sci. Rep. 2017, 7, 15130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andralojc, K.; Srinivas, M.; Brom, M.; Joosten, L.; de Vries, I.J.M.; Eizirik, D.L.; Boerman, O.C.; Meda, P.; Gotthardt, M. Obstacles on the Way to the Clinical Visualisation of Beta Cells: Looking for the Aeneas of Molecular Imaging to Navigate between Scylla and Charybdis. Diabetologia 2012, 55, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Md Moin, A.S.; Dhawan, S.; Shieh, C.; Butler, P.C.; Cory, M.; Butler, A.E. Increased Hormone-Negative Endocrine Cells in the Pancreas in Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3487–3496. [Google Scholar] [CrossRef] [Green Version]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieira, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The Inactivation of Arx in Pancreatic α-Cells Triggers Their Neogenesis and Conversion into Functional β-Like Cells. PLoS Genet. 2013, 9, e1003934. [Google Scholar] [CrossRef]
- Demine, S.; Garcia Ribeiro, R.; Thevenet, J.; Marselli, L.; Marchetti, P.; Pattou, F.; Kerr-Conte, J.; Devoogdt, N.; Eizirik, D.L. A Nanobody-Based Nuclear Imaging Tracer Targeting Dipeptidyl Peptidase 6 to Determine the Mass of Human Beta Cell Grafts in Mice. Diabetologia 2020, 63, 825–836. [Google Scholar] [CrossRef]
- Zheng, F.; Sparkes, A.; De Baetselier, P.; Schoonooghe, S.; Stijlemans, B.; Muyldermans, S.; Flamand, V.; Van Ginderachter, J.A.; Devoogdt, N.; Raes, G.; et al. Molecular Imaging with Kupffer Cell-Targeting Nanobodies for Diagnosis and Prognosis in Mouse Models of Liver Pathogenesis. Mol. Imaging Biol. 2017, 19, 49–58. [Google Scholar] [CrossRef]
- Fallatah, H.I. Noninvasive Biomarkers of Liver Fibrosis: An Overview. Adv. Hepatol. 2014, 2014, 1–15. [Google Scholar] [CrossRef]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver Inflammation Abrogates Immunological Tolerance Induced by Kupffer Cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Devoogdt, N.; Sparkes, A.; Morias, Y.; Abels, C.; Stijlemans, B.; Lahoutte, T.; Muyldermans, S.; De Baetselier, P.; Schoonooghe, S.; et al. Monitoring Liver Macrophages Using Nanobodies Targeting Vsig4: Concanavalin A Induced Acute Hepatitis as Paradigm. Immunobiology 2015, 220, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Allison, J.P. To Be or Not to Be B7. J. Clin. Investig. 2006, 116, 2590–2593. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone Marrow-Derived Monocytes Give Rise to Self-Renewing and Fully Differentiated Kupffer Cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Ramadori, P.; Weiskirchen, R.; Trebicka, J.; Streetz, K. Mouse Models of Metabolic Liver Injury. Lab. Anim. 2015, 49, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Lambert, B.; Cybulla, M.; Weiner, S.M.; Van De Wiele, C.; Ham, H.; Dierckx, R.A.; Otte, A. Renal Toxicity after Radionuclide Therapy. Radiat. Res. 2004, 161, 607–611. [Google Scholar] [CrossRef]
- Vegt, E.; de Jong, M.; Wetzels, J.F.M.; Masereeuw, R.; Melis, M.; Oyen, W.J.G.; Gotthardt, M.; Boerman, O.C. Renal Toxicity of Radiolabeled Peptides and Antibody Fragments: Mechanisms, Impact on Radionuclide Therapy, and Strategies for Prevention. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- van Eerd, J.E.M.; Vegt, E.; Wetzels, J.F.M.; Russel, F.G.M.; Masereeuw, R.; Corstens, F.H.M.; Oyen, W.J.G.; Boerman, O.C. Gelatin-Based Plasma Expander Effectively Reduces Renal Uptake of 111In-Octreotide in Mice and Rats. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2006, 47, 528–533. [Google Scholar]
- Vegt, E.; Wetzels, J.F.M.; Russel, F.G.M.; Masereeuw, R.; Boerman, O.C.; van Eerd, J.E.; Corstens, F.H.M.; Oyen, W.J.G. Renal Uptake of Radiolabeled Octreotide in Human Subjects Is Efficiently Inhibited by Succinylated Gelatin. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2006, 47, 432–436. [Google Scholar]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.J.; Kuhn, B.T.; Bräuer, P.; Newstead, S.; Dawson, R.J.P.; Geertsma, E.R.; Seeger, M.A. Generation of Synthetic Nanobodies against Delicate Proteins. Nat. Protoc. 2020, 15, 1707–1741. [Google Scholar] [CrossRef]
- Zhou, Z.; Vaidyanathan, G.; McDougald, D.; Kang, C.M.; Balyasnikova, I.; Devoogdt, N.; Ta, A.N.; McNaughton, B.R.; Zalutsky, M.R. Fluorine-18 Labeling of the HER2-Targeting Single-Domain Antibody 2Rs15d Using a Residualizing Label and Preclinical Evaluation. Mol. Imaging Biol. 2017, 19, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Schoonooghe, S.; Laoui, D.; Houbracken, I.; Waelput, W.; Breckpot, K.; Bouwens, L.; Lahoutte, T.; Baetselier, P.D.; Raes, G.; et al. Nanobody-Based Targeting of the Macrophage Mannose Receptor for Effective In Vivo Imaging of Tumor-Associated Macrophages. Cancer Res. 2012, 72, 4165–4177. [Google Scholar] [CrossRef] [Green Version]
- Groeve, K.D.; Deschacht, N.; Koninck, C.D.; Caveliers, V.; Lahoutte, T.; Devoogdt, N.; Muyldermans, S.; Baetselier, P.D.; Raes, G. Nanobodies as Tools for In Vivo Imaging of Specific Immune Cell Types. J. Nucl. Med. 2010, 51, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Broos, K.; Lecocq, Q.; Xavier, C.; Bridoux, J.; Nguyen, T.T.; Corthals, J.; Schoonooghe, S.; Lion, E.; Raes, G.; Keyaerts, M.; et al. Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent. Cancers 2019, 11, 872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Jiang, L.; Li, K.; Li, H.; Lv, G.; Lin, J.; Qiu, L. Immuno-PET Imaging of 68Ga-Labeled Nanobody Nb109 for Dynamic Monitoring the PD-L1 Expression in Cancers. Cancer Immunol. Immunother. CII 2021. [Google Scholar] [CrossRef] [PubMed]
- Sun Yoo, J.; Lee, J.; Ho Jung, J.; Seok Moon, B.; Kim, S.; Chul Lee, B.; Eun Kim, S. SPECT/CT Imaging of High-Risk Atherosclerotic Plaques Using Integrin-Binding RGD Dimer Peptides. Sci. Rep. 2015, 5, 11752. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, R.; Goel, S.; Cai, W. Nanobody: The “Magic Bullet” for Molecular Imaging? Theranostics 2014, 4, 386–398. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berland, L.; Kim, L.; Abousaway, O.; Mines, A.; Mishra, S.; Clark, L.; Hofman, P.; Rashidian, M. Nanobodies for Medical Imaging: About Ready for Prime Time? Biomolecules 2021, 11, 637. https://doi.org/10.3390/biom11050637
Berland L, Kim L, Abousaway O, Mines A, Mishra S, Clark L, Hofman P, Rashidian M. Nanobodies for Medical Imaging: About Ready for Prime Time? Biomolecules. 2021; 11(5):637. https://doi.org/10.3390/biom11050637
Chicago/Turabian StyleBerland, Léa, Lauren Kim, Omar Abousaway, Andrea Mines, Shruti Mishra, Louise Clark, Paul Hofman, and Mohammad Rashidian. 2021. "Nanobodies for Medical Imaging: About Ready for Prime Time?" Biomolecules 11, no. 5: 637. https://doi.org/10.3390/biom11050637
APA StyleBerland, L., Kim, L., Abousaway, O., Mines, A., Mishra, S., Clark, L., Hofman, P., & Rashidian, M. (2021). Nanobodies for Medical Imaging: About Ready for Prime Time? Biomolecules, 11(5), 637. https://doi.org/10.3390/biom11050637