
Modulation of the EMT/MET Process by E-Cadherin in Airway Epithelia Stress Injury


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model of Injury Repair
2.2. Measurement of Airway Function
2.3. Histology, H&E, and Immunochemistry
2.4. Cell Culture and Model of Injury Repair
2.5. Overexpression Plasmid Synthesis and Transfection
2.6. Total RNA Extraction and Real-Time PCR
2.7. Western Blotting
2.8. Immunofluorescence
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Statistical Analysis
3. Results
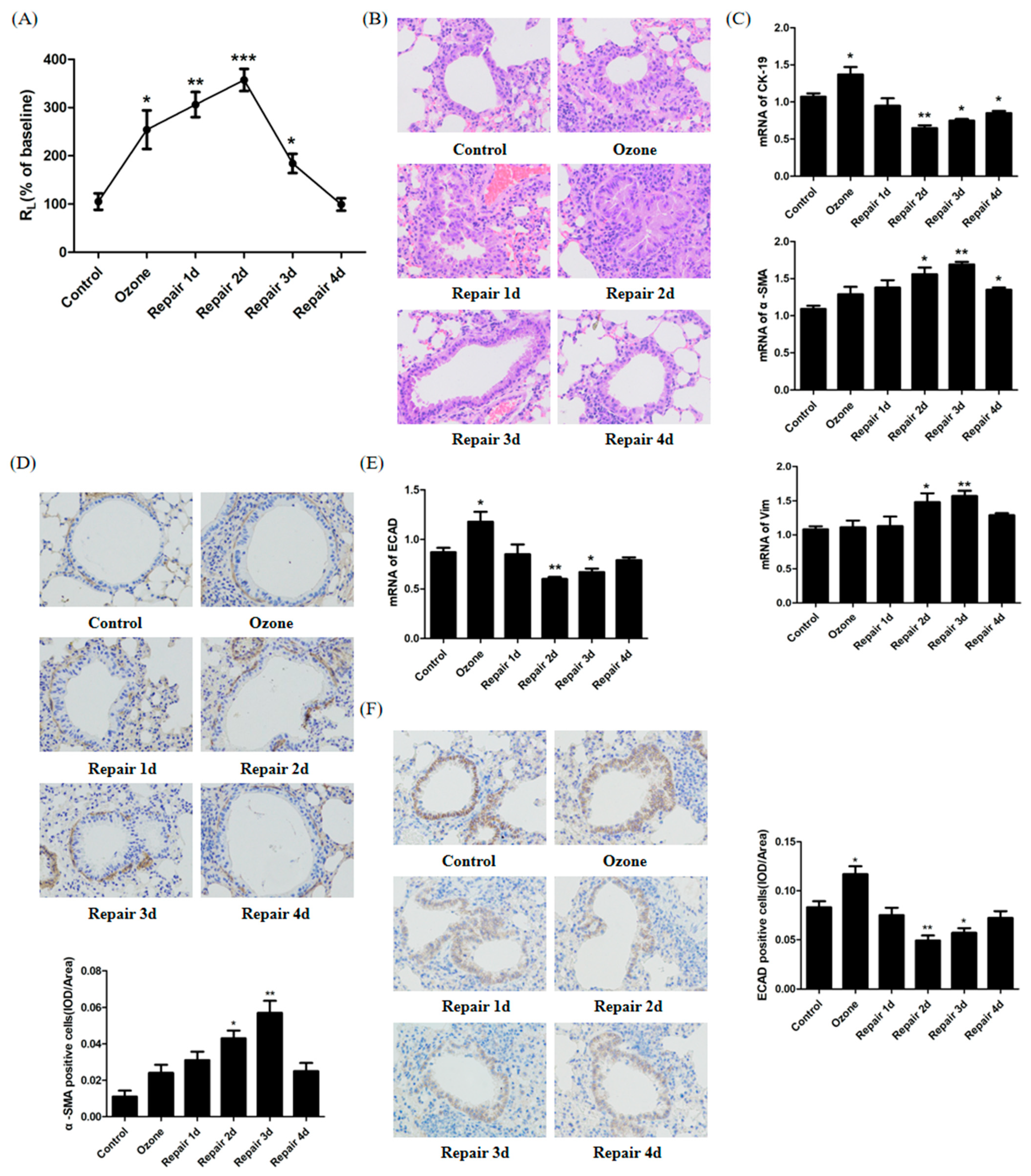
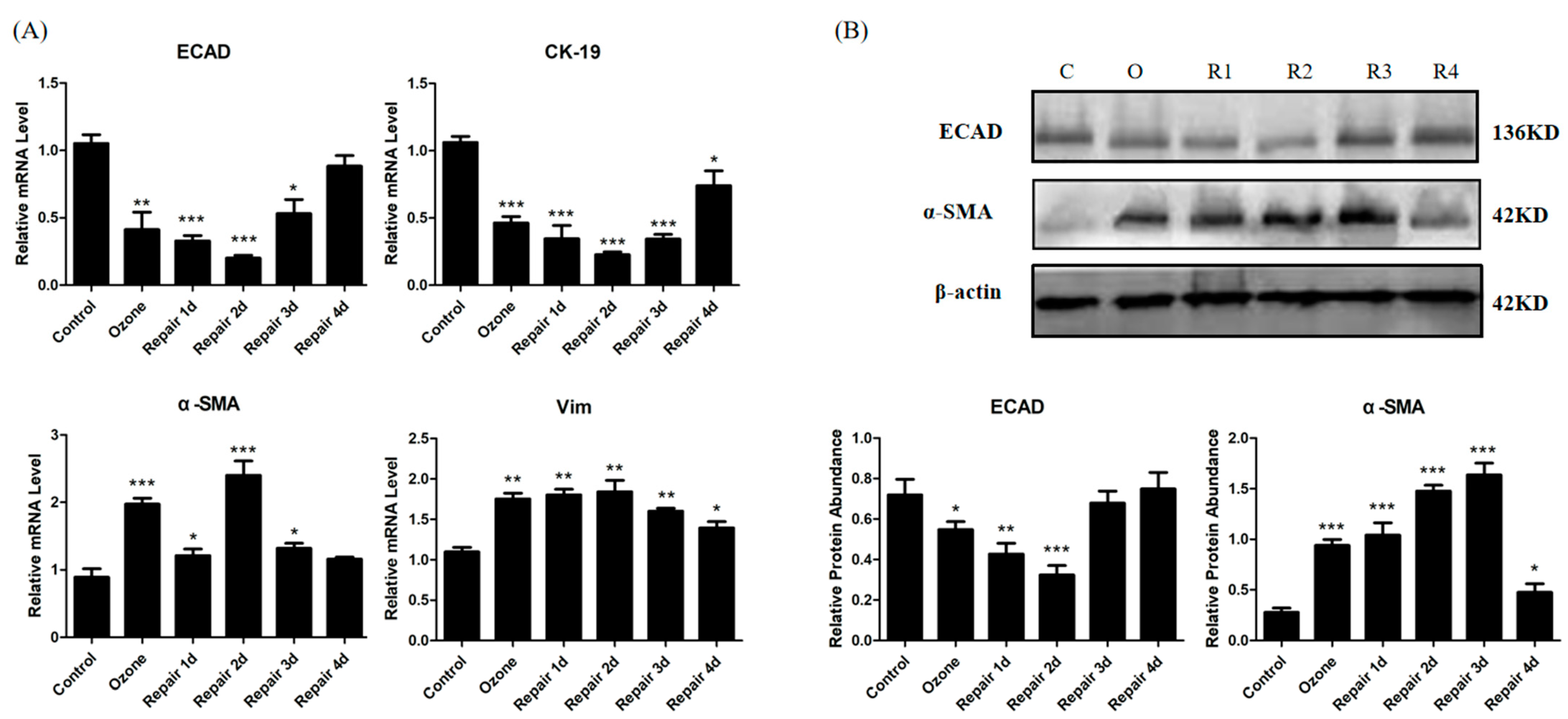
3.1. ECAD Correlates with EMT/MET and Airway Epithelia Injury Repair in the Ozone-Stressed Mice Model
3.2. EMT/MET Features in Bronchial Epithelial Cells in Ozone-Induced Injury Repair
3.3. ECAD Overexpression Represses EMT Features and Promotes MET Features in Ozone-Stressed HBECs
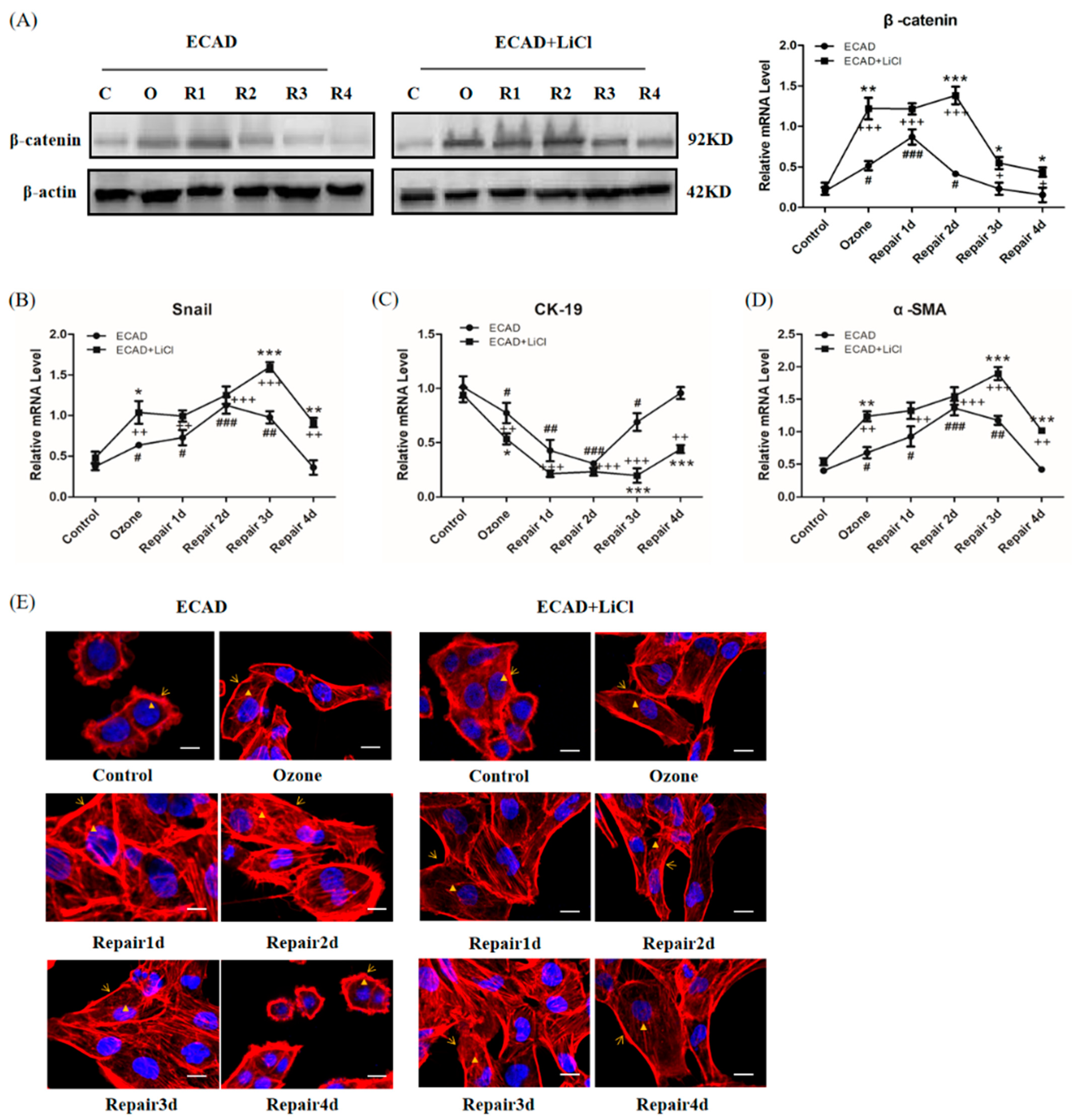
3.4. The Effects of ECAD on EMT/MET Is Mediated by TGFβ1/Snail Signaling
3.5. Role of β-catenin in ECAD Inhibition Effect on TGFβ1/Snail Signaling
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Boulet, L.P. Airway remodeling in asthma: Update on mechanisms and therapeutic approaches. Curr. Opin. Pulm. Med. 2018, 24, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [Green Version]
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Faura Tellez, G.; Willemse, B.W.; Brouwer, U.; Nijboer-Brinksma, S.; Vandepoele, K.; Noordhoek, J.A.; Heijink, I.; de Vries, M.; Smithers, N.P.; Postma, D.S.; et al. Protocadherin-1 Localization and Cell-Adhesion Function in Airway Epithelial Cells in Asthma. PLoS ONE 2016, 11, e0163967. [Google Scholar]
- Post, S.; Heijink, I.H.; Hesse, L.; Koo, H.K.; Shaheen, F.; Fouadi, M.; Kuchibhotla, V.N.S.; Lambrecht, B.N.; Van Oosterhout, A.J.M.; Hackett, T.L.; et al. Characterization of a lung epithelium specific E-cadherin knock-out model: Implications for obstructive lung pathology. Sci. Rep. 2018, 8, 13275. [Google Scholar] [CrossRef] [Green Version]
- Aresu, L.; Rastaldi, M.P.; Pregel, P.; Valenza, F.; Radaelli, E.; Scanziani, E.; Castagnaro, M. Dog as model for down-expression of E-cadherin and beta-catenin in tubular epithelial cells in renal fibrosis. Virchows Arch. 2008, 453, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Kim, Y.W.; Han, C.Y.; Kim, E.H.; Anderson, R.A.; Lee, Y.S.; Lee, C.H.; Hwang, S.J.; Kim, S.G. E-cadherin antagonizes transforming growth factor beta1 gene induction in hepatic stellate cells by inhibiting RhoA-dependent Smad3 phosphorylation. Hepatology 2010, 52, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Satow, R.; Kato, A.; Tamura, M.; Murayama, Y.; Saya, H.; Kojima, H.; Nagano, T.; Okabe, T.; Fukami, K. Identification of novel small compounds that restore E-cadherin expression and inhibit tumor cell motility and invasiveness. Biochem. Pharmacol. 2013, 86, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Seidel, B.; Braeg, S.; Adler, G.; Wedlich, D.; Menke, A. E- and N-cadherin differ with respect to their associated p120ctn isoforms and their ability to suppress invasive growth in pancreatic cancer cells. Oncogene 2004, 23, 5532–5542. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Xiang, Y.; Liu, H.J.; Gao, G.; Howard, S.T.; Zhu, X.L.; Qin, X.Q. Involvement of integrin beta4 in ozone stress-induced airway hyperresponsiveness. Biochem. Biophys. Res. Commun. 2010, 397, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, X.; Zheng, D.; Zhang, D.; Huang, S.; Zhang, X.; Ai, F.; Wang, X.; Ma, J.; Xiong, W.; et al. Dynamic changes of peritoneal macrophages and subpopulations during ulcerative colitis to metastasis of colorectal carcinoma in a mouse model. Inflamm. Res. 2013, 62, 669–680. [Google Scholar] [CrossRef]
- Gruenert, D.C.; Finkbeiner, W.E.; Widdicombe, J.H. Culture and transformation of human airway epithelial cells. Am. J. Physiol. 1995, 268, L347–L360. [Google Scholar] [CrossRef]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef]
- Tan, M.; Liu, C.; Huang, W.; Deng, L.; Qin, X.; Xiang, Y. CTNNAL1 inhibits ozone-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Exp. Physiol. 2018, 103, 1157–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.R.; Oslund, K.L.; Hyde, D.M.; Miller, L.A.; Van Winkle, L.S.; Schelegle, E.S. Ozone-induced airway epithelial cell death, the neurokinin-1 receptor pathway, and the postnatal developing lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L471–L481. [Google Scholar] [CrossRef] [Green Version]
- Harkema, J.R.; Wagner, J.G. Innate Lymphoid Cell-Dependent Airway Epithelial and Inflammatory Responses to Inhaled Ozone: A New Paradigm in Pathogenesis. Toxicol. Pathol. 2019, 47, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.A. Wound epithelialization: Accelerating the pace of discovery. J. Investig. Dermatol. 2003, 121, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Perotin, J.M.; Adam, D.; Vella-Boucaud, J.; Delepine, G.; Sandu, S.; Jonvel, A.C.; Prevost, A.; Berthiot, G.; Pison, C.; Lebargy, F.; et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir. Res. 2014, 15, 151. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Pelaez, L.M.; Abraham, T.; Herrera, A.M.; Correa, M.A.; Ortega, J.E.; Pare, P.D.; Seow, C.Y. Vitronectin expression in the airways of subjects with asthma and chronic obstructive pulmonary disease. PLoS ONE 2015, 10, e0119717. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D.M.; Stow, J.L. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 2004, 14, 427–434. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/beta-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peplow, P.V.; Chatterjee, M.P. A review of the influence of growth factors and cytokines in in vitro human keratinocyte migration. Cytokine 2013, 62, 1–21. [Google Scholar] [CrossRef]
- Chiba, T.; Ishisaki, A.; Kyakumoto, S.; Shibata, T.; Yamada, H.; Kamo, M. Transforming growth factor-beta1 suppresses bone morphogenetic protein-2-induced mesenchymal-epithelial transition in HSC-4 human oral squamous cell carcinoma cells via Smad1/5/9 pathway suppression. Oncol. Rep. 2017, 37, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.L.; Mainez, J.; Vega, S.; Sancho, P.; Murillo, M.M.; de Frutos, C.A.; Del Castillo, G.; Lopez-Blau, C.; Fabregat, I.; Nieto, M.A. Snail1 suppresses TGF-beta-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J. Cell Sci. 2010, 123, 3467–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol. 2018, 1095, 101–110. [Google Scholar]
- Wang, W.; Liu, F.; Wang, C.; Wang, C.; Tang, Y.; Jiang, Z. Glutathione S-transferase A1 mediates nicotine-induced lung cancer cell metastasis by promoting epithelial-mesenchymal transition. Exp. Ther. Med. 2017, 14, 1783–1788. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target/Control Gene | Primer Sequences |
|---|---|
| Mouse ECAD | Forward 5′-ACCGGAAGTGACTCGAAATGATGT-3′ |
| Reverse 5′-CTTCAGAACCACTGCCCTCGTAAT-3′ | |
| Mouse CK-19 | Forward 5′-GGTTCAGTACGCATTGGGTCA-3′ |
| Reverse 5′-CGGAGGACGAGGTCACGAA-3′ | |
| Mouse α-SMA | Forward 5′-CCCAGATTATGTTTGAGACC-3′ |
| Reverse 5′- TCCAGAGTCCAGCACAATAC-3′ | |
| Mouse Vim | Forward 5′-AAGCACCCTGCAGTCATTCA-3′ |
| Reverse 5′- AGGCTTGGAAACGTCCACAT-3′ | |
| Mouse β-actin | Forward 5′-TTGCAGCTCCTTCGTTGCC-3′ |
| Reverse 5′-GACCCATTCCCACCATCACA-3′ | |
| Human β-actin | Forward 5′-TTCCAGCCTTCCTTCCTGGG-3′ |
| Reverse 5′-TTGCGCTCAGGAGGAGCAAT-3′ | |
| Human ECAD | Forward 5′-TCCAGGAACCTCTGTGATGGA-3′ |
| Reverse 5′-ACTCTCTCGGTCCAGCCCA-3′ | |
| Human CK-19 | Forward 5′-TTTGAGACGGAACAGGCTCT-3′ |
| Reverse 5′-AGGCTTGGAAACGTCCACAT-3′ | |
| Human α-SMA | Forward 5′-GTGTTGCCCCTGAAGAGCAT-3′ |
| Reverse 5′-GCTGGGACATTGAAAGTCTCA-3′ | |
| Human Vim | Forward 5′-TGGACCAGCTAACCAACGAC-3′ |
| Reverse 5′-GCCAGAGACGCATTGTCAAC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, L.; Luo, H.; Huang, W.; Zhang, J.; Wu, D.; Wang, J.; Pi, J.; Liu, C.; Qu, X.; Liu, H.; et al. Modulation of the EMT/MET Process by E-Cadherin in Airway Epithelia Stress Injury. Biomolecules 2021, 11, 669. https://doi.org/10.3390/biom11050669
Han L, Luo H, Huang W, Zhang J, Wu D, Wang J, Pi J, Liu C, Qu X, Liu H, et al. Modulation of the EMT/MET Process by E-Cadherin in Airway Epithelia Stress Injury. Biomolecules. 2021; 11(5):669. https://doi.org/10.3390/biom11050669
Chicago/Turabian StyleHan, Li, Huaiqing Luo, Wenjie Huang, Jiang Zhang, Di Wu, Jinmei Wang, Jiao Pi, Chi Liu, Xiangping Qu, Huijun Liu, and et al. 2021. "Modulation of the EMT/MET Process by E-Cadherin in Airway Epithelia Stress Injury" Biomolecules 11, no. 5: 669. https://doi.org/10.3390/biom11050669