
Computational Study on Selective PDE9 Inhibitors on PDE9-Mg/Mg, PDE9-Zn/Mg, and PDE9-Zn/Zn Systems

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Ligand Topology
2.3. Molecular Docking
2.4. Molecular Dynamics Simulations
2.5. MM/PBSA Binding Free Energy Calculation
3. Results and Discussion
3.1. Docking of PDE-9 Inhibitors with PDE-9
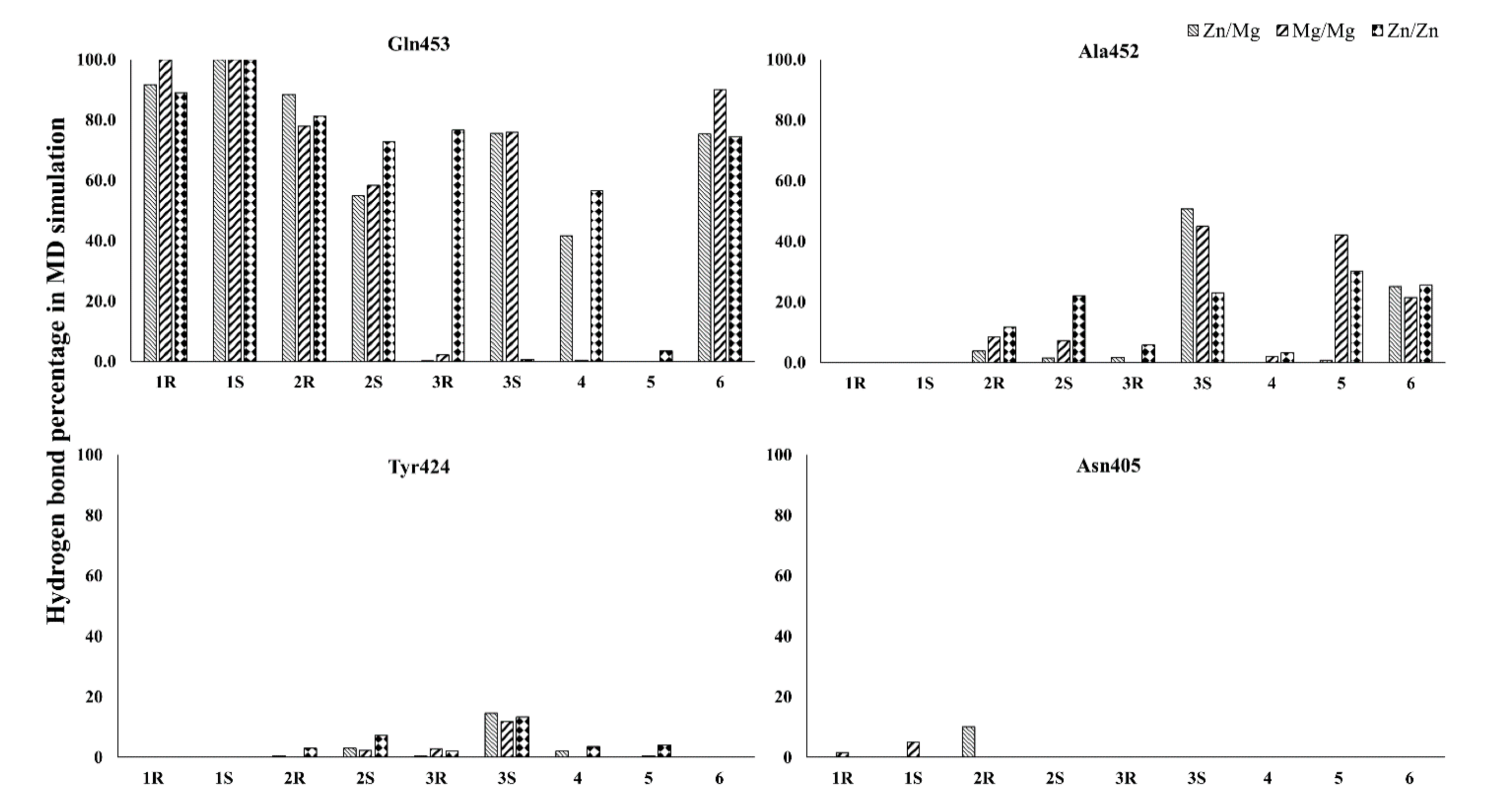
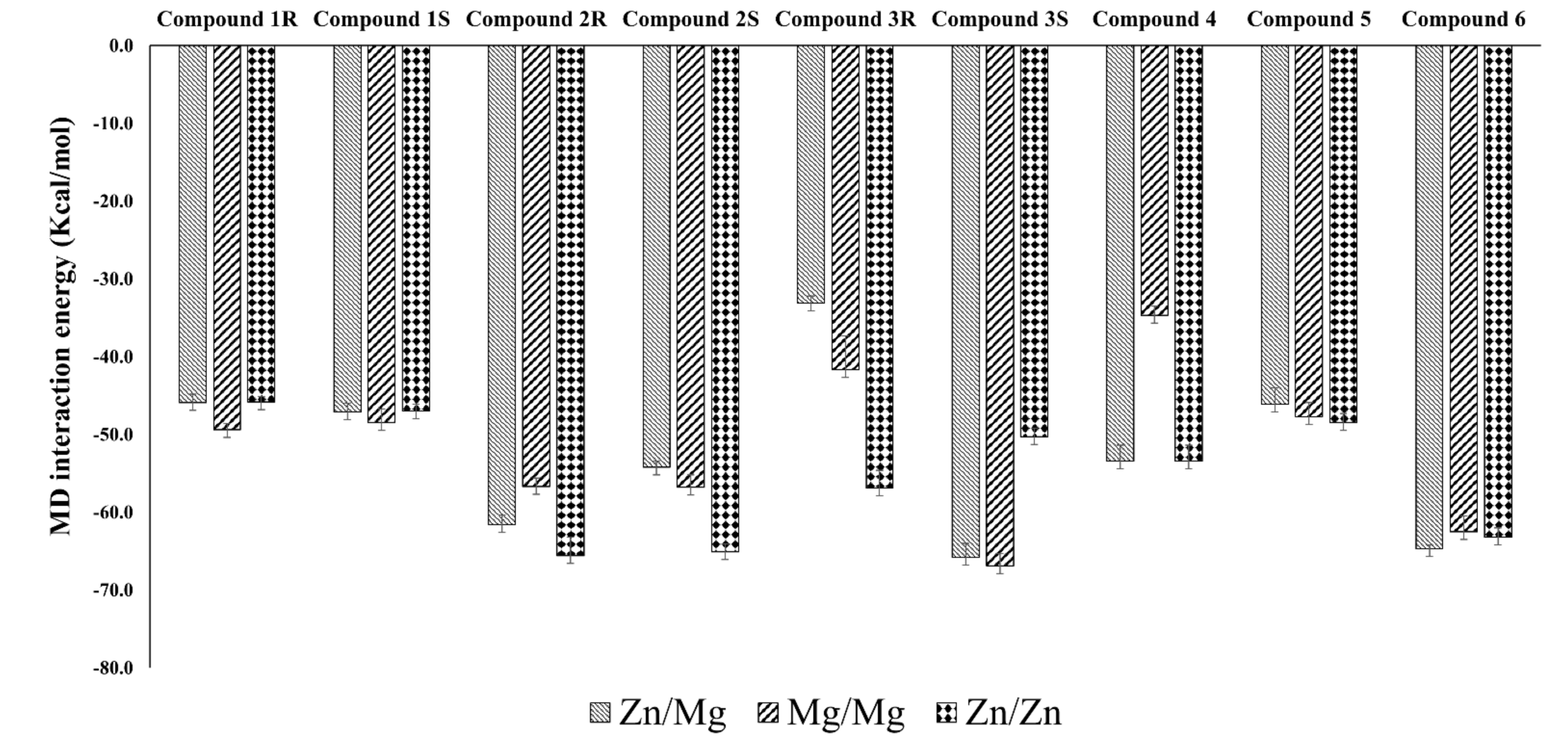
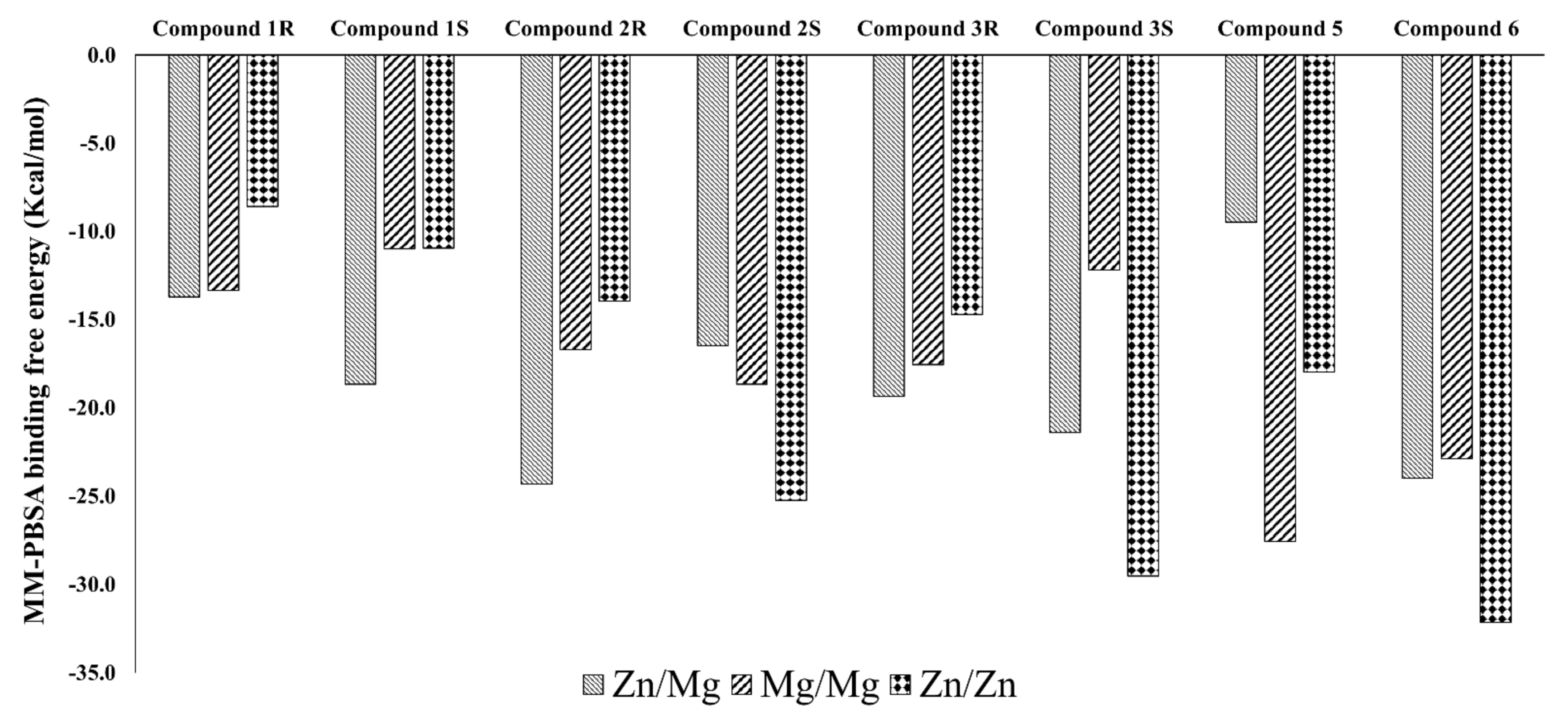
3.2. Interaction Details Based on the Simulation Results
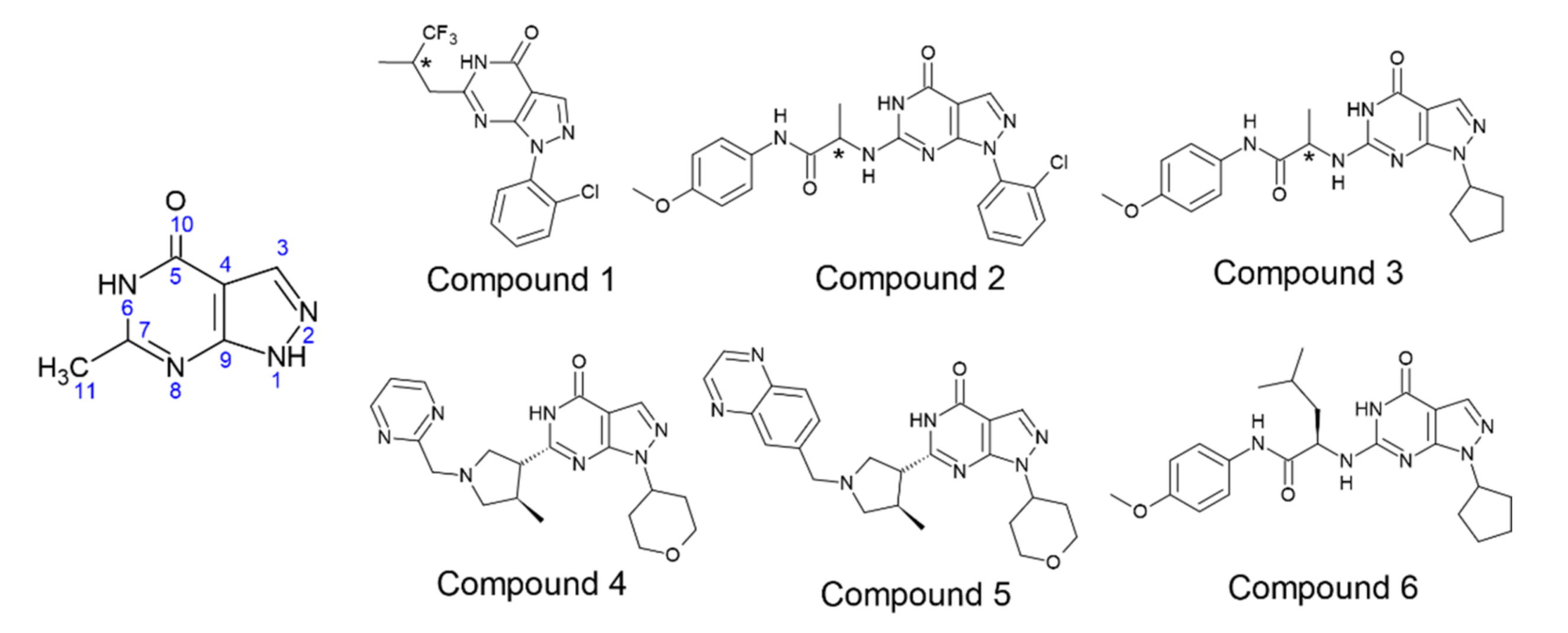
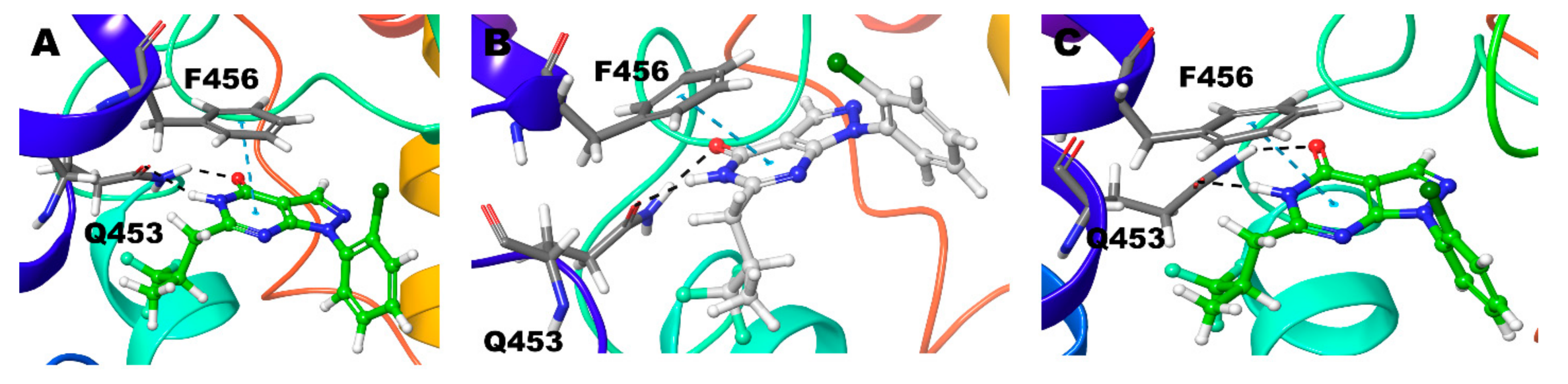
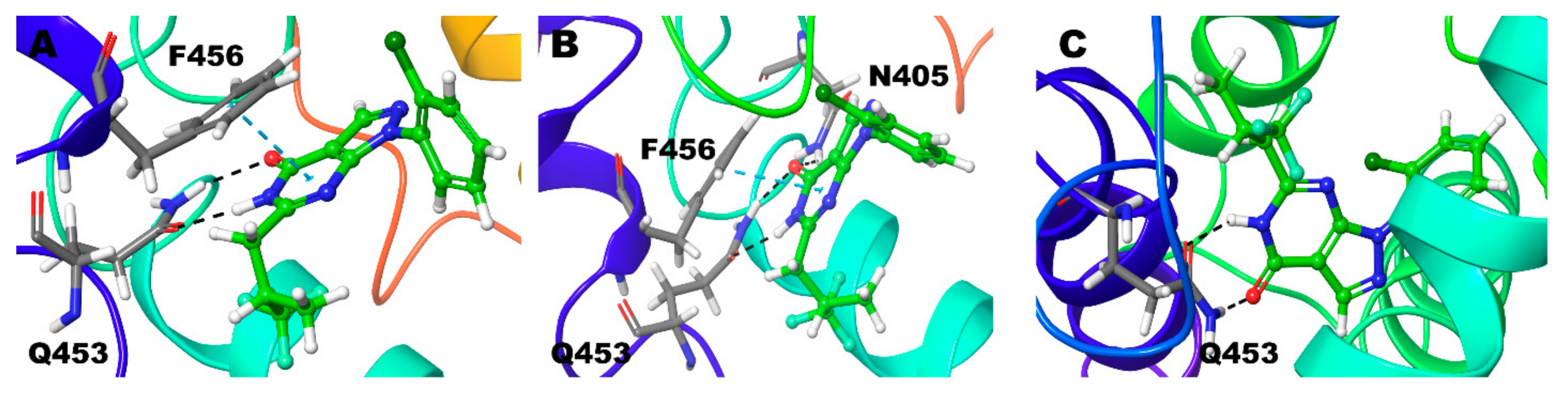
3.2.1. Compound 1 (BAY73-6691 -R and S) in PDE-9 Zn/Mg, PDE-9 Mg/Mg, and PDE-9 Zn/Zn
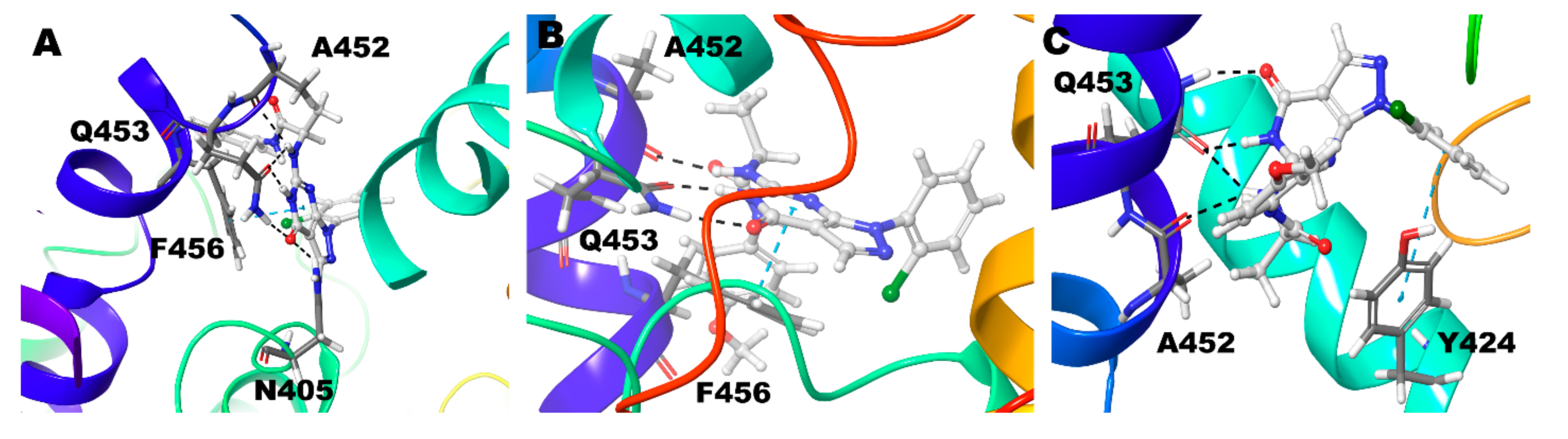
3.2.2. Compound 2 (28R and 28S) in PDE-9 Zn/Mg, PDE-9 Mg/Mg, and PDE-9 Zn/Zn
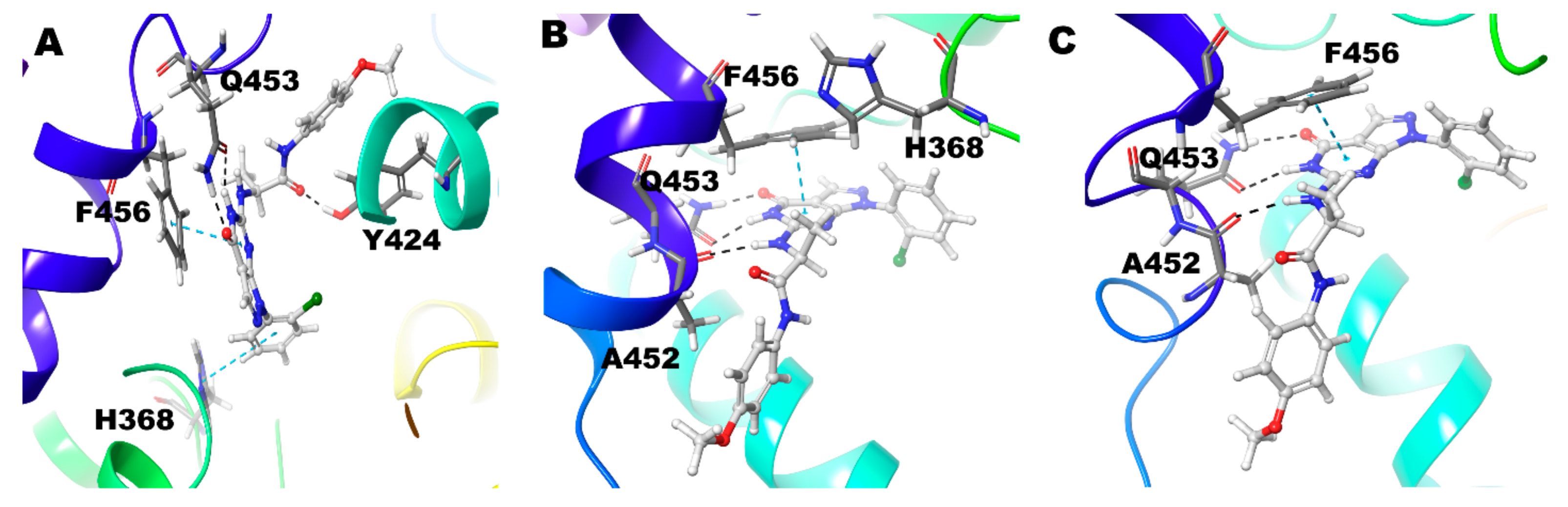
3.2.3. Compound 3 (3R and 3S) in PDE-9 Zn/Mg, PDE-9 Mg/Mg, and PDE-9 Zn/Zn
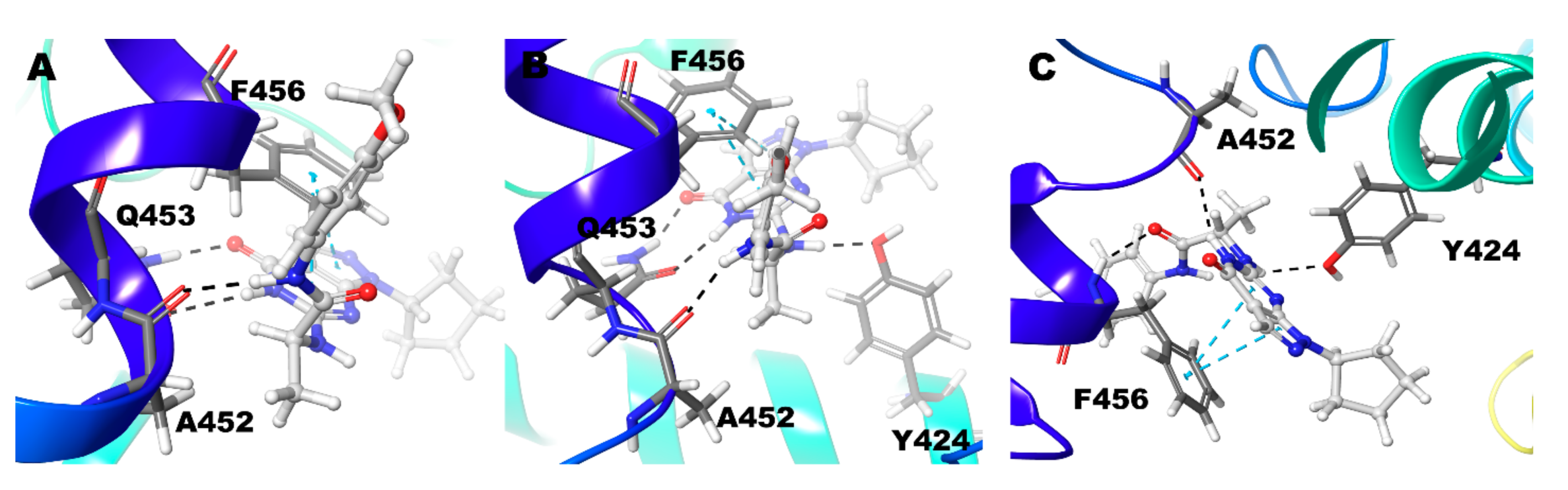
3.2.4. Compound 4 (PF-04447943) in PDE-9 Zn/Mg, PDE-9 Mg/Mg, and PDE-9 Zn/Zn
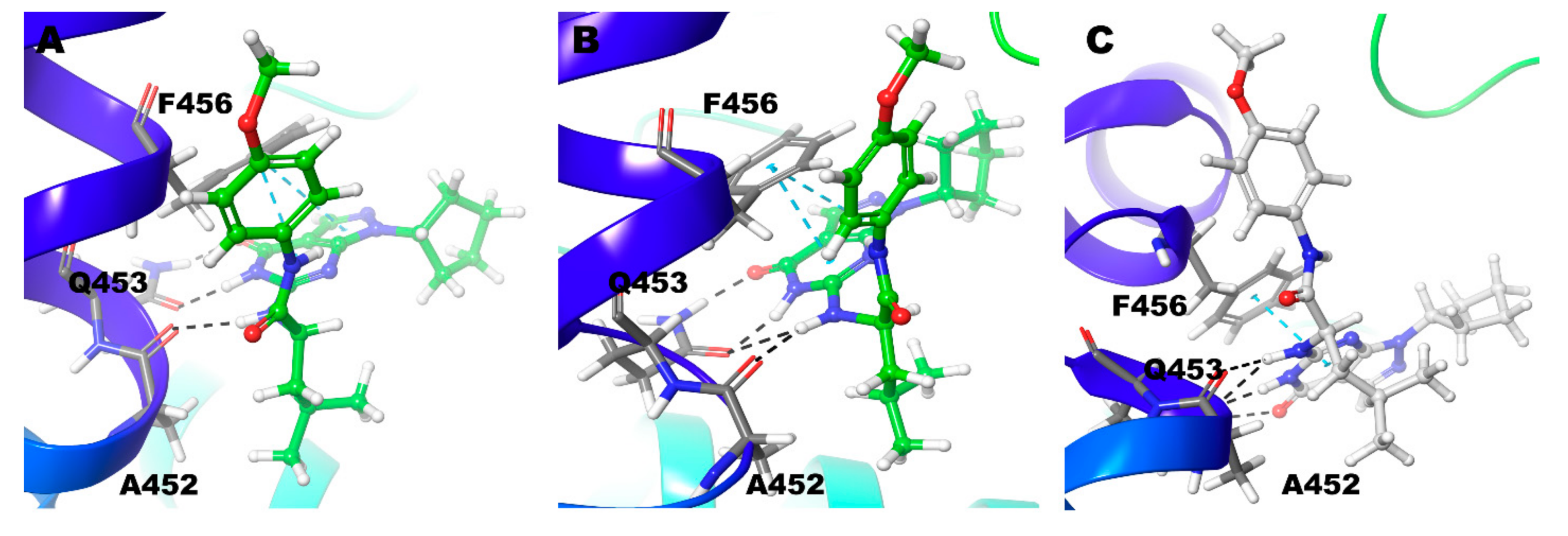
3.2.5. Compound 5 (PF-4181366) in PDE-9 Zn/Mg, PDE-9 Mg/Mg and PDE-9 Zn/Zn
3.2.6. Compound 6 (4r) in PDE-9 Zn/Mg, PDE-9 Mg/Mg and PDE-9 Zn/Zn
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat. Rev. Drug. Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küthe, A.; Wiedenroth, A.; Mägert, H.J.; Uckert, S.; Forssmann, W.G.; Stief, C.G.; Jonas, U. Expression of Different Phosphodiesterase Genes in Human Cavernous Smooth Muscle. J. Urol. 2001, 165, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, P.; Egan, R.W.; Billah, M.M. Identification and Characterization of a New Human Type 9 CGMP-Specific Phosphodiesterase Splice Variant (PDE9A5). Differential Tissue Distribution and Subcellular Localization of PDE9A Variants. Gene 2003, 314, 15–27. [Google Scholar] [CrossRef]
- Almeida, C.B.; Traina, F.; Lanaro, C.; Canalli, A.A.; Saad, S.T.O.; Costa, F.F.; Conran, N. High Expression of the CGMP-Specific Phosphodiesterase, PDE9A, in Sickle Cell Disease (SCD) and the Effects of Its Inhibition in Erythroid Cells and SCD Neutrophils. Br. J. Haematol. 2008, 142, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, S.; Nakano, Y.; Masuda, M.; Ono, K.; Miki, Y.; Shibahara, Y.; Sasano, H. Phosphodiesterase Type 9 (PDE9) in the Human Lower Urinary Tract: An Immunohistochemical Study. BJU Int. 2012, 109, 934–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, M.; Jin, S.L. The Molecular Biology of Cyclic Nucleotide Phosphodiesterases. Prog Nucleic. Acid. Res. Mol. Biol. 1999, 63, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic Nucleotide Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and Physiology of Cyclic Nucleotide Phosphodiesterases: Essential Components in Cyclic Nucleotide Signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian Cyclic Nucleotide Phosphodiesterases: Molecular Mechanisms and Physiological Functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [Green Version]
- Keravis, T.; Lugnier, C. Cyclic Nucleotide Phosphodiesterase (PDE) Isozymes as Targets of the Intracellular Signalling Network: Benefits of PDE Inhibitors in Various Diseases and Perspectives for Future Therapeutic Developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [Green Version]
- Fryburg, D.; Gibbs, E. Treatment of Insulin Resistance Syndrome and Type 2 Diabetes with Pde9 Inhibitors 2003. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2003037432 (accessed on 23 March 2021).
- US20170173018 Substituted Imidazo [1,5-a]Pyrazines as PDE9 Inhibitors. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=US199414325&docAn=15452827 (accessed on 23 March 2021).
- Wunder, F.; Tersteegen, A.; Rebmann, A.; Erb, C.; Fahrig, T.; Hendrix, M. Characterization of the First Potent and Selective PDE9 Inhibitor Using a CGMP Reporter Cell Line. Mol. Pharmacol. 2005, 68, 1775–1781. [Google Scholar] [CrossRef]
- Verhoest, P.R.; Proulx-Lafrance, C.; Corman, M.; Chenard, L.; Helal, C.J.; Hou, X.; Kleiman, R.; Liu, S.; Marr, E.; Menniti, F.S.; et al. Identification of a Brain Penetrant PDE9A Inhibitor Utilizing Prospective Design and Chemical Enablement as a Rapid Lead Optimization Strategy. J. Med. Chem. 2009, 52, 7946–7949. [Google Scholar] [CrossRef]
- Verhoest, P.R.; Fonseca, K.R.; Hou, X.; Proulx-Lafrance, C.; Corman, M.; Helal, C.J.; Claffey, M.M.; Tuttle, J.B.; Coffman, K.J.; Liu, S.; et al. Design and Discovery of 6-[(3S,4S)-4-Methyl-1-(Pyrimidin-2-Ylmethyl)Pyrrolidin-3-Yl]-1-(Tetrahydro-2H-Pyran-4-Yl)-1,5-Dihydro-4H-Pyrazolo[3,4-d]Pyrimidin-4-One (PF-04447943), a Selective Brain Penetrant PDE9A Inhibitor for the Treatment of Cognitive Disorders. J. Med. Chem. 2012, 55, 9045–9054. [Google Scholar] [CrossRef]
- Meng, F.; Hou, J.; Shao, Y.-X.; Wu, P.-Y.; Huang, M.; Zhu, X.; Cai, Y.; Li, Z.; Xu, J.; Liu, P.; et al. Structure-Based Discovery of Highly Selective Phosphodiesterase-9A Inhibitors and Implications for Inhibitor Design. J. Med. Chem. 2012, 55, 8549–8558. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Huang, M.; Cui, W.; Feng, L.-J.; Wu, Y.; Cai, Y.; Li, Z.; Zhu, X.; Liu, P.; Wan, Y.; et al. Discovery of a Phosphodiesterase 9A Inhibitor as a Potential Hypoglycemic Agent. J. Med. Chem. 2014, 57, 10304–10313. [Google Scholar] [CrossRef] [Green Version]
- van der Staay, F.J.; Rutten, K.; Bärfacker, L.; DeVry, J.; Erb, C.; Heckroth, H.; Karthaus, D.; Tersteegen, A.; van Kampen, M.; Blokland, A.; et al. The Novel Selective PDE9 Inhibitor BAY 73-6691 Improves Learning and Memory in Rodents. Neuropharmacology 2008, 55, 908–918. [Google Scholar] [CrossRef] [Green Version]
- da Silva, F.H.; Pereira, M.N.; Franco-Penteado, C.F.; De Nucci, G.; Antunes, E.; Claudino, M.A. Phosphodiesterase-9 (PDE9) Inhibition with BAY 73-6691 Increases Corpus Cavernosum Relaxations Mediated by Nitric Oxide–Cyclic GMP Pathway in Mice. Int. J. Impot. Res. 2013, 25, 69–73. [Google Scholar] [CrossRef]
- Kleiman, R.J.; Chapin, D.S.; Christoffersen, C.; Freeman, J.; Fonseca, K.R.; Geoghegan, K.F.; Grimwood, S.; Guanowsky, V.; Hajós, M.; Harms, J.F.; et al. Phosphodiesterase 9A Regulates Central CGMP and Modulates Responses to Cholinergic and Monoaminergic Perturbation in Vivo. J. Pharmacol. Exp. Ther 2012, 341, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Hutson, P.H.; Finger, E.N.; Magliaro, B.C.; Smith, S.M.; Converso, A.; Sanderson, P.E.; Mullins, D.; Hyde, L.A.; Eschle, B.K.; Turnbull, Z.; et al. The Selective Phosphodiesterase 9 (PDE9) Inhibitor PF-04447943 (6-[(3S,4S)-4-Methyl-1-(Pyrimidin-2-Ylmethyl)Pyrrolidin-3-Yl]-1-(Tetrahydro-2H-Pyran-4-Yl)-1,5-Dihydro-4H-Pyrazolo[3,4-d]Pyrimidin-4-One) Enhances Synaptic Plasticity and Cognitive Function in Rodents. Neuropharmacology 2011, 61, 665–676. [Google Scholar] [CrossRef]
- Charnigo, R.J.; Beidler, D.; Rybin, D.; Pittman, D.D.; Tan, B.; Howard, J.; Michelson, A.D.; Frelinger, A.L.; Clarke, N. PF-04447943, a Phosphodiesterase 9A Inhibitor, in Stable Sickle Cell Disease Patients: A Phase Ib Randomized, Placebo-Controlled Study. Clin. Transl. Sci. 2019, 12, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Harms, J.F.; Menniti, F.S.; Schmidt, C.J. Phosphodiesterase 9A in Brain Regulates CGMP Signaling Independent of Nitric-Oxide. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Protein Data Bank|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/28/1/235/2384399 (accessed on 25 March 2021).
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. On The Berendsen Thermostat. Mol. Simul. 1994, 13, 177–187. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, D.; Kumar, V.; Naumann, M.; Stein, M. Activation and Selectivity of OTUB-1 and OTUB-2 Deubiquitinylases. J. Biol. Chem. 2020, 295, 6972–6982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivakumar, D.; Gorai, B.; Sivaraman, T. Screening Efficient BH3-Mimetics to HBcl-B by Means of Peptidodynmimetic Method. Mol. BioSyst. 2013, 9, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Enantiomer | Docking Score (kcal/mol) | Interacting Residues * |
|---|---|---|---|
| Compound 1 | R | −9.5 | F251, Q453, F456 |
| Compound 1 | S | −9.5 | F251, Q453, F456 |
| Compound 2 | R | −11.3 | F251, A452, Q453, F456 |
| Compound 2 | S | −10.9 | A452, Q453, F456 |
| Compound 3 | R | −10.7 | F251, Y424, Q453, F456 |
| Compound 3 | S | −10.3 | A452, Q453, F456 |
| Compound 4 | n/a | −10.7 | F251, Q453, F456 |
| Compound 5 | n/a | −12.0 | F251, Q453, F456 |
| Compound 6 | n/a | −9.3 | A452, Q453, F456 |
| Compound ID/Enantiomer | Metal System | Hydrogen Bond Interactions | Hydrophobic Contacts |
|---|---|---|---|
| Compound 1 (R) | Zn/Mg | Q453 | F456 |
| Mg/Mg | Q453 | F456 | |
| Zn/Zn | Q453 | F456 | |
| Compound 1 (S) | Zn/Mg | Q453 | F456 |
| Mg/Mg | Q453, N405 | F456 | |
| Zn/Zn | Q453 | No Interactions | |
| Compound 2 (R) | Zn/Mg | Q453, A452, N405 | F456 |
| Mg/Mg | Q453, A452 | F456 | |
| Zn/Zn | Q453, A452 | Y424 | |
| Compound 2 (S) | Zn/Mg | Q453, Y424 | F456, H368 |
| Mg/Mg | Q453, A452 | F456, H368 | |
| Zn/Zn | Q453, A452 | F456 | |
| Compound 3 (R) | Zn/Mg | T302 | Y424, F441 |
| Mg/Mg | No Interactions | No Interactions | |
| Zn/Zn | Q453 | F456 | |
| Compound 3 (S) | Zn/Mg | Q453, A452 | F456 |
| Mg/Mg | Q453, A452, Y424 | F456 | |
| Zn/Zn | Y424, A452, F456 | F456 | |
| Compound 4 | Zn/Mg | Q453 | F456, F441 |
| Mg/Mg | No Interactions | F456 | |
| Zn/Zn | Q453 | F456 | |
| Compound 5 | Zn/Mg | No Interactions | No Interactions |
| Mg/Mg | A452 | F441 | |
| Zn/Zn | A452 | F456, F441 | |
| Compound 6 | Zn/Mg | Q453, A452 | F456 |
| Mg/Mg | Q453, A452 | F456 | |
| Zn/Zn | Q453, A452 | F456 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivakumar, D.; Mudedla, S.; Jang, S.; Kim, H.; Park, H.; Choi, Y.; Oh, J.; Wu, S. Computational Study on Selective PDE9 Inhibitors on PDE9-Mg/Mg, PDE9-Zn/Mg, and PDE9-Zn/Zn Systems. Biomolecules 2021, 11, 709. https://doi.org/10.3390/biom11050709
Sivakumar D, Mudedla S, Jang S, Kim H, Park H, Choi Y, Oh J, Wu S. Computational Study on Selective PDE9 Inhibitors on PDE9-Mg/Mg, PDE9-Zn/Mg, and PDE9-Zn/Zn Systems. Biomolecules. 2021; 11(5):709. https://doi.org/10.3390/biom11050709
Chicago/Turabian StyleSivakumar, Dakshinamurthy, Sathishkumar Mudedla, Seonghun Jang, Hyunjun Kim, Hyunjin Park, Yonwon Choi, Joongyo Oh, and Sangwook Wu. 2021. "Computational Study on Selective PDE9 Inhibitors on PDE9-Mg/Mg, PDE9-Zn/Mg, and PDE9-Zn/Zn Systems" Biomolecules 11, no. 5: 709. https://doi.org/10.3390/biom11050709
APA StyleSivakumar, D., Mudedla, S., Jang, S., Kim, H., Park, H., Choi, Y., Oh, J., & Wu, S. (2021). Computational Study on Selective PDE9 Inhibitors on PDE9-Mg/Mg, PDE9-Zn/Mg, and PDE9-Zn/Zn Systems. Biomolecules, 11(5), 709. https://doi.org/10.3390/biom11050709