
Transcriptome and Metabolome Reveal Salt-Stress Responses of Leaf Tissues from Dendrobium officinale

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Design
2.2. RNA Extraction and Sequencing
2.3. Quantitative Reverse Transcription-PCR
2.4. Nontargeted Metabolic Profiling
2.5. Measurement of Anthocyanidin, Total Flavonoid, and JA Content
3. Results
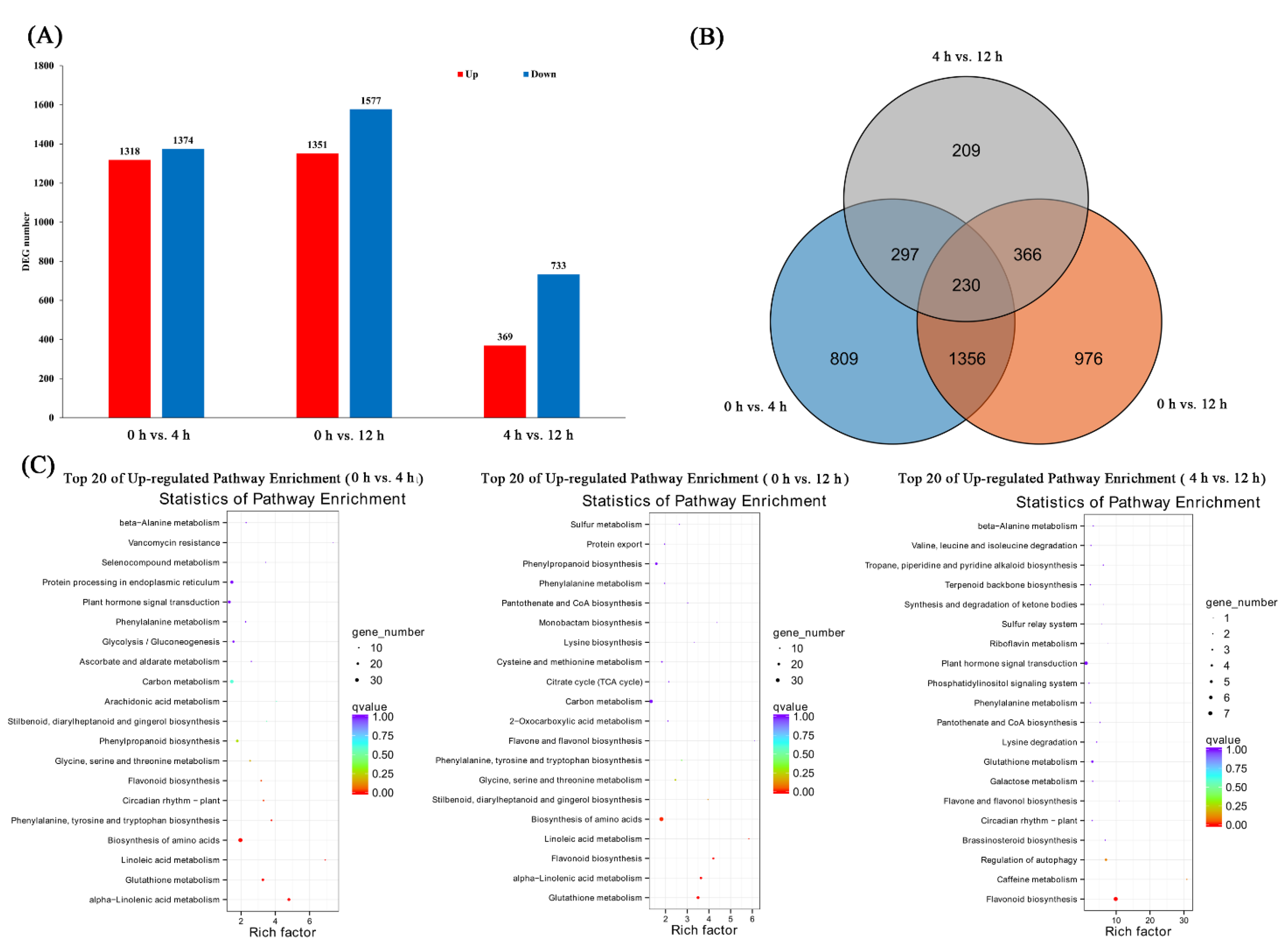
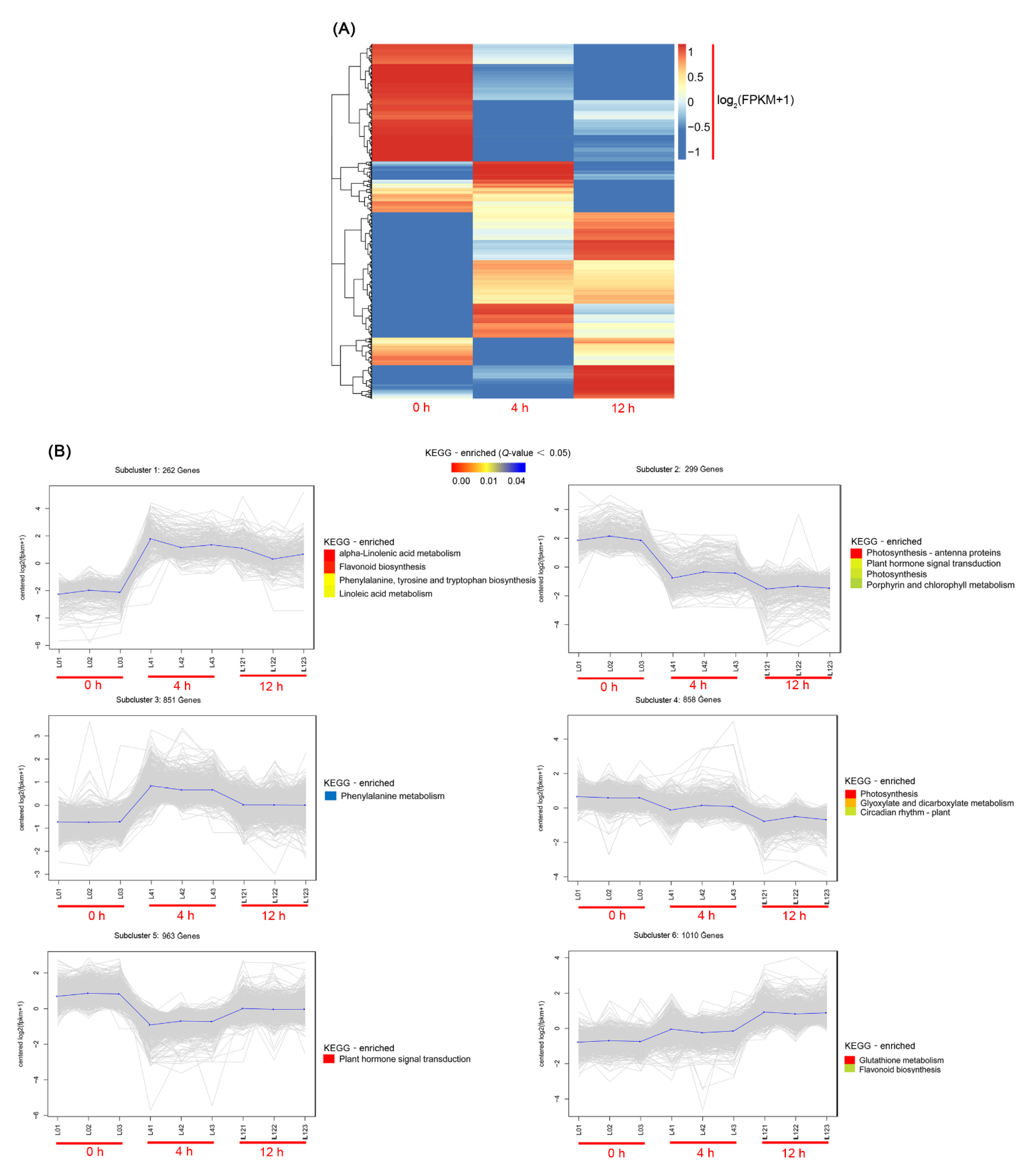
3.1. Transcriptional Profiling
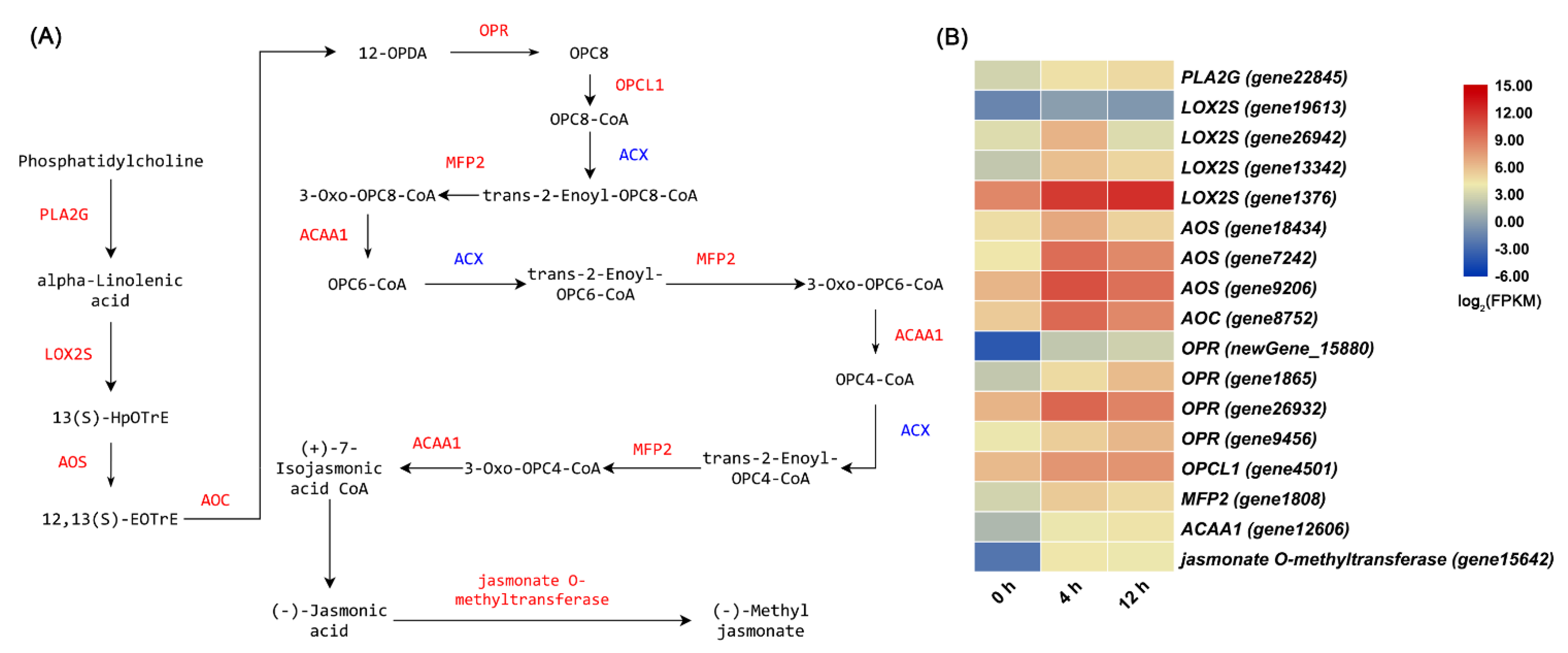
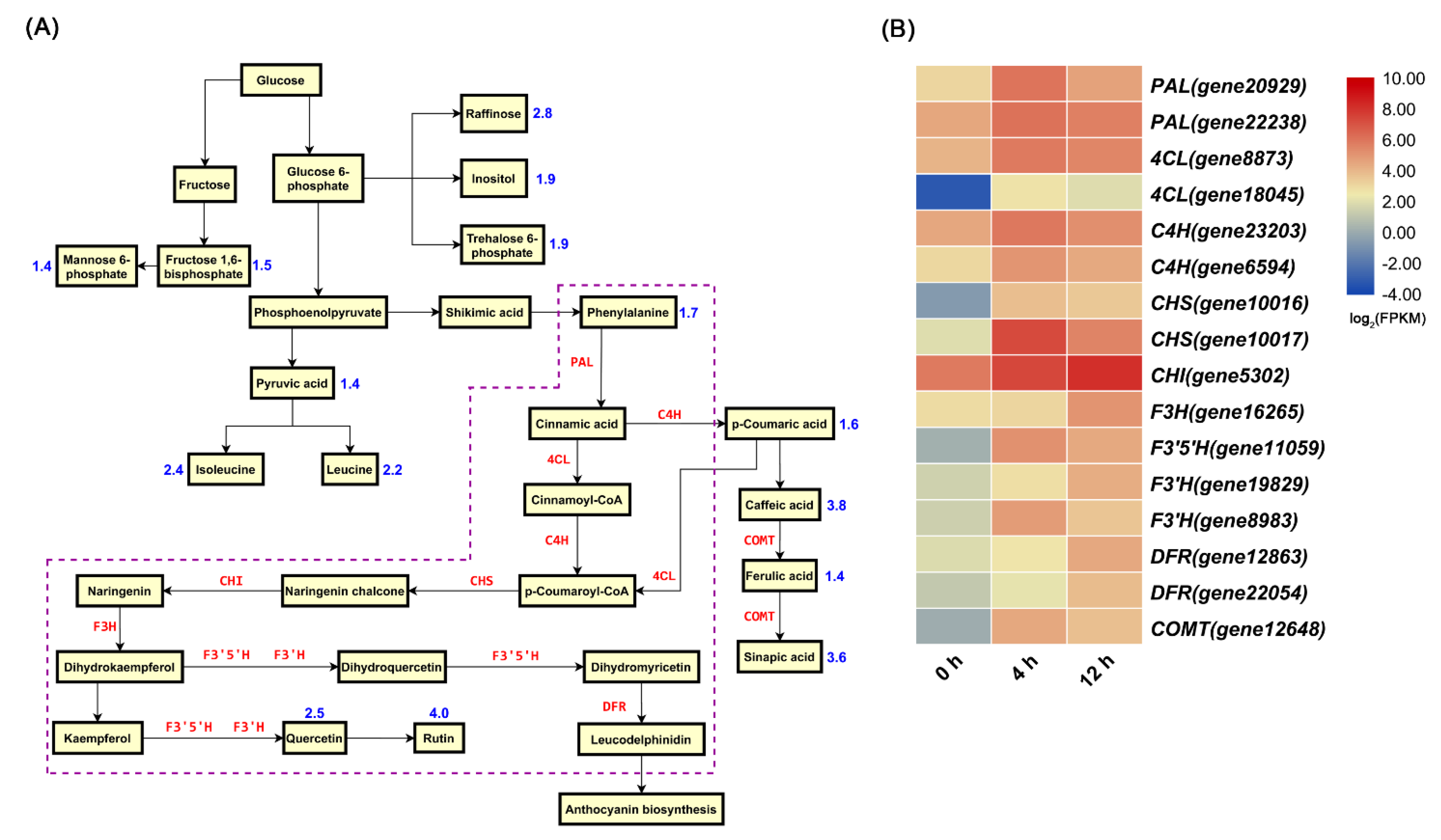
3.2. Metabolomic Profiling and DEGs in Related Pathways
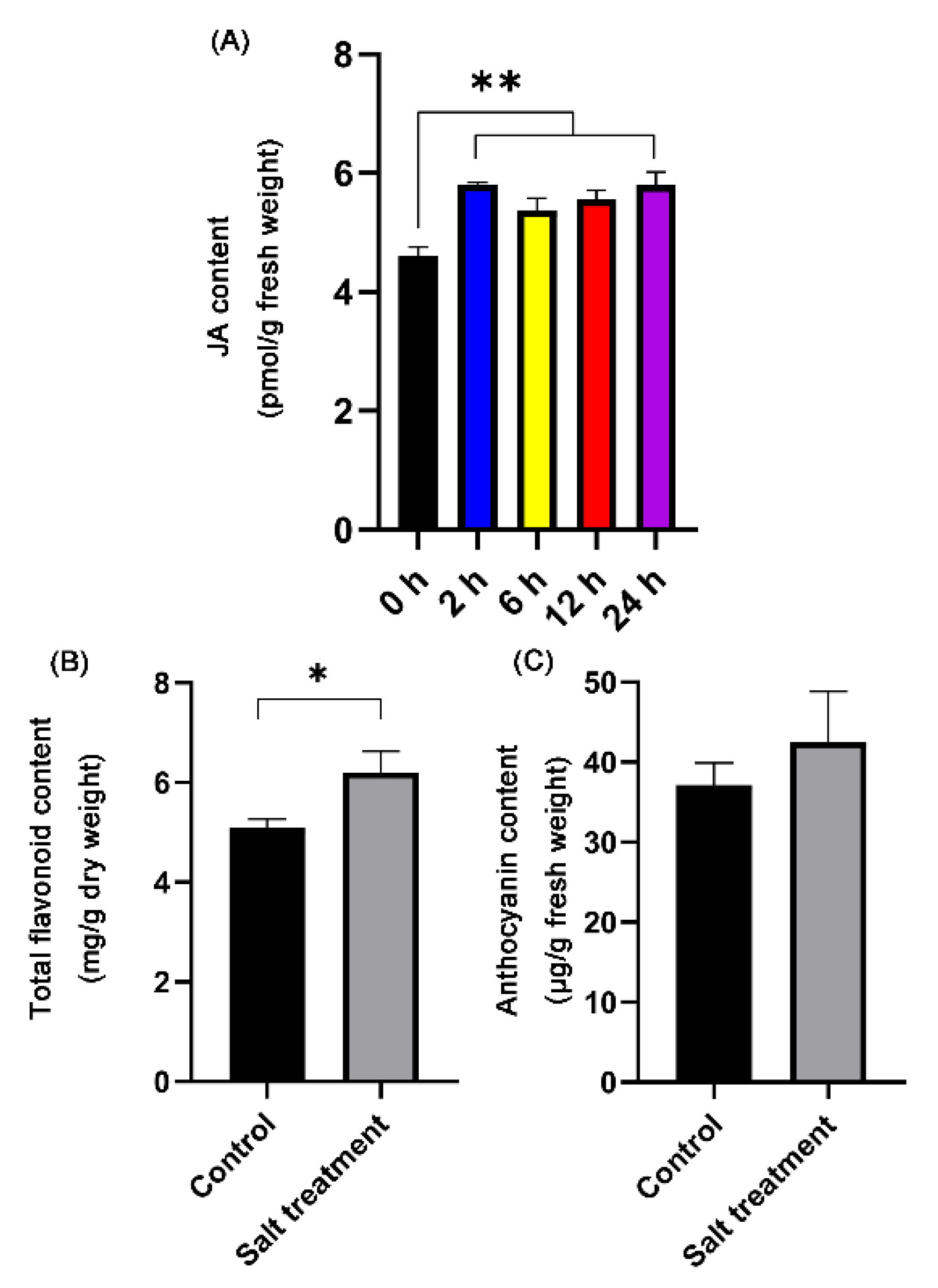
3.3. Salt Stress Enhanced the Contents of Total Flavonoids, Anthocyanidin, and JA
4. Discussion
4.1. The Molecular Mechanisms of D. officinale Leaves in Response to Salt Stress
4.2. Salt Stress Induces the Accumulation of Bioactive Compounds in D. officinale Leaves
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lam, Y.; Ng, T.B.; Yao, R.M.; Shi, J.; Xu, K.; Sze, S.C.W.; Zhang, K.Y. Evaluation of chemical constituents and important mechanism of pharmacological biology in Dendrobium plants. Evid. Based Complement. Altern. Med. 2015, 2015, 841752. [Google Scholar]
- Li, X.; Ding, X.; Chu, B.; Zhou, Q.; Ding, G.; Gu, S. Genetic diversity analysis and conservation of the endangered Chinese endemic herb Dendrobium officinale Kimura et Migo (Orchidaceae) based on AFLP. Genetica 2008, 133, 159–166. [Google Scholar]
- Tang, H.; Zhao, T.; Sheng, Y.; Zheng, T.; Fu, L.; Zhang, Y. Dendrobium officinale Kimura et Migo: A review on its ethnopharmacology, phytochemistry, pharmacology, and industrialization. Evid. Based Complement. Altern. Med. 2017, 2017, 7436259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, C.; Song, C.; Zheng, S.; Zhu, Y.; Jin, Q.; Cai, Y.; Lin, Y. Metabolic profiling of Dendrobium officinale in response to precursors and methyl jasmonate. Int. J. Mol. Sci. 2018, 19, 728. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Guo, H.; Chen, H.; Shi, Y.; Meng, Y.; Lu, J.; Feng, S.; Wang, H. Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci. Rep. 2017, 7, 187. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Yu, D.; Xue, J.; Lu, J.; Feng, S.; Shen, C.; Wang, H. A transcriptome-wide, organ-specific regulatory map of Dendrobium officinale, an important traditional Chinese orchid herb. Sci. Rep. 2016, 6, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, T.; Liu, J.; Wong, J.; Ye, X.; Sze, S.C.W.; Tong, Y.; Zhang, K.Y. Review of research on Dendrobium, a prized folk medicine. Appl. Microbiol. Biotechnol. 2012, 93, 1795–1803. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Liu, J.; Liang, J.; Si, J.; Wu, S. Dendrobium officinale leaves as a new antioxidant source. J. Funct. Foods 2017, 37, 400–415. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Ma, B.; Gao, Q.; Du, H.; Han, Y.; Li, Y.; Cao, Y.; Qi, M.; Zhu, Y. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol. Plant 2017, 10, 1224–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, S.; Cornish, K.; Kumar, S. Salinity tolerance in plants: Breeding and genetic engineering. Aust. J. Crop Sci. 2012, 6, 1337–1348. [Google Scholar]
- Tuteja, N. Mechanisms of high salinity tolerance in plants. Methods Enzymol. 2007, 428, 419–438. [Google Scholar]
- Gupta, B.; Huang, B. Mechanism of salinity tolerance in plants: Physiological, biochemical, and molecular characterization. Int. J. Genomics 2014, 2014, 701596. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Pandeyrai, S. Updates on artemisinin: An insight to mode of actions and strategies for enhanced global production. Protoplasma 2016, 253, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Supatida, A.; Pawanrat, K.; Piyaklao, T.; Parson, S. Physiological responses of potted Dendrobium orchid to salinity stress. Hortic. Environ. Biotechnol. 2018, 59, 491–498. [Google Scholar]
- Yang, D.; Du, X.; Yang, Z.; Liang, Z.; Guo, Z.; Liu, Y. Transcriptomics, proteomics, and metabolomics to reveal mechanisms underlying plant secondary metabolism. Eng. Life Sci. 2014, 14, 456–466. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.Q.; Liu, K.W.; Li, Z.; Lohaus, R.; Hsiao, Y.Y.; Niu, S.; Wang, J.Y.; Lin, Y.C.; Chen, L.J.; Yoshida, K.; et al. The Apostasia genome and the evolution of orchids. Nature 2017, 549, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Liu, X.; Silva, J.A.T.D.; Liu, N.; Duan, J. Transcriptome sequencing and metabolite profiling analyses provide comprehensive insight into molecular mechanisms of flower development in Dendrobium officinale (Orchidaceae). Plant Mol. Biol. 2020, 104, 529–548. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Wang, J.; Xian, B.; Tang, X.; Pei, J. Transcriptome analysis of flavonoid biosynthesis in safflower flowers grown under different light intensities. PeerJ 2020, 8, e8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulaev, V.; Cortes, D.; Miller, G.; Mittler, R. Metabolomics for plant stress response. Physiol. Plant. 2008, 132, 199–208. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B.; Mohanty, P. Investigations on the antioxidative defence responses to NaCl stress in a mangrove, Bruguiera parviflora: Differential regulations of isoforms of some antioxidative enzymes. Plant Growth Regul. 2004, 42, 213–226. [Google Scholar] [CrossRef]
- Fernandez, O.; Bethencourt, L.; Quero, A.; Sangwan, R.S.; Clement, C. Trehalose and plant stress responses: Friend or foe? Trends Plant Sci. 2010, 15, 409–417. [Google Scholar] [CrossRef]
- Paul, M.J.; Primavesi, L.F.; Jhurreea, D.; Zhang, Y. Trehalose metabolism and signaling. Annu. Rev. Plant Biol. 2008, 59, 417–441. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, Y.; Liu, Y.; Li, X.; Hao, G.; Han, Q.; Dirk, L.M.A.; Downie, A.B.; Ruan, Y.L.; Wang, J.; et al. Raffinose synthase enhances drought tolerance through raffinose synthesis or galactinol hydrolysis in maize and Arabidopsis plants. J. Biol. Chem. 2020, 295, 8064–8077. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, L.; Ye, Z.; Li, H. Ascorbic acid accumulation is transcriptionally modulated in high-pigment-1tomato fruit. Plant Mol. Biol. Rep. 2014, 32, 52–61. [Google Scholar] [CrossRef]
- Smirnoff, N.; Conklin, P.L.; Loewus, F.A. Biosynthesis of ascorbic acid in plants: A renaissance. Annu. Rev. Plant Physiol. 2001, 52, 437–467. [Google Scholar] [CrossRef]
- He, C.; Yu, Z.; Teixeira da Silva, J.A.; Zhang, J.; Liu, X.; Wang, X.; Zhang, X.; Zeng, S.; Wu, K.; Tan, J. DoGMP1 from Dendrobium officinale contributes to mannose content of water-soluble polysaccharides and plays a role in salt stress response. Sci. Rep. 2017, 7, 41010. [Google Scholar] [CrossRef] [Green Version]
- Nazar, R.; Umar, S.; Khan, N.A. Exogenous salicylic acid improves photosynthesis and growth through increase in ascorbate-glutathione metabolism and S assimilation in mustard under salt stress. Plant Signal. Behav. 2015, 10, e1003751. [Google Scholar] [CrossRef] [Green Version]
- Badawi, G.H.; Kawano, N.; Yamauchi, Y.; Shimada, E.; Sasaki, R.; Kubo, A.; Tanaka, K. Over-expression of ascorbate peroxidase in tobacco chloroplasts enhances the tolerance to salt stress and water deficit. Physiol. Plant. 2004, 121, 231–238. [Google Scholar] [CrossRef]
- Ji, W.; Zhu, Y.; Li, Y.; Yang, L.; Zhao, X.; Cai, H.; Bai, X. Over-expression of a glutathione S-transferase gene, GsGST, from wild soybean (Glycine soja) enhances drought and salt tolerance in transgenic tobacco. Biotechnol. Lett. 2010, 32, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Zhai, C.Z.; Zhao, L.; Yin, L.J.; Chen, M.; Wang, Q.Y.; Li, L.C.; Xu, Z.S.; Ma, Y.Z. Two wheat glutathione peroxidase genes whose products are located in chloroplasts improve salt and H2O2 tolerances in Arabidopsis. PLoS ONE 2013, 8, e73989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.M.; Lin, W.R.; Kao, Y.T.; Hsu, Y.T.; Yeh, C.H.; Hong, C.Y.; Kao, C.H. Identification and characterization of a novel chloroplast/mitochondria co-localized glutathione reductase 3 involved in salt stress response in rice. Plant Mol. Biol. 2013, 83, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.M.; Lin, W.R.; Kao, C.H.; Hong, C.Y. Gene knockout of glutathione reductase 3 results in increased sensitivity to salt stress in rice. Plant Mol. Biol. 2015, 87, 555–564. [Google Scholar] [CrossRef]
- Pauwels, L.; Morreel, K.; De Witte, E.; Lammertyn, F.; Van Montagu, M.; Boerjan, W.; Inze, D.; Goossens, A. Mapping methyl jasmonate-mediated transcriptional reprogramming of metabolism and cell cycle progression in cultured Arabidopsis cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1380–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Jin, M.; Paek, K.; Piao, X.; Lian, M. An efficient strategy for enhancement of bioactive compounds by protocorm-like body culture of Dendrobium candidum. Ind. Crop. Prod. 2016, 84, 121–130. [Google Scholar] [CrossRef]
- Yuan, Z.Q.; Zhang, J.; Liu, T. Enhancement of polysaccharides accumulation in Dendrobium officinale by exogenously applied methyl jasmonate. Biol. Plant. 2017, 61, 438–444. [Google Scholar] [CrossRef]
- Glauser, G.; Dubugnon, L.; Mousavi, S.A.R.; Rudaz, S.; Wolfender, J.L.; Farmer, E.E. Velocity estimates for signal propagation leading to systemic jasmonic acid accumulation in wounded Arabidopsis. J. Biol. Chem. 2009, 284, 34506–34513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thines, B.; Katsir, L.; Melotto, M.; Niu, Y.; Mandaokar, A.; Liu, G.; Nomura, K.; He, S.Y.; Howe, G.A.; Browse, J. JAZ repressor proteins are targets of the SCFCOI1 complex during jasmonate signalling. Nature 2007, 448, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wood, K.V.; Morgan, J.A. Metabolic engineering of the phenylpropanoid pathway in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2005, 71, 2962–2969. [Google Scholar] [CrossRef] [Green Version]
- Ravaglia, D.; Espley, R.V.; Henrykirk, R.A.; Andreotti, C.; Ziosi, V.; Hellens, R.P.; Costa, G.; Allan, A.C. Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol. 2013, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.M.; Schwinn, K.E. Transcriptional regulation of secondary metabolism. Funct. Plant Biol. 2003, 30, 913–925. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Yu, Z.; Zeng, D.; Si, C.; Zhao, C.; Wang, H.; Li, C.; He, C.; Duan, J. Transcriptome and Metabolome Reveal Salt-Stress Responses of Leaf Tissues from Dendrobium officinale. Biomolecules 2021, 11, 736. https://doi.org/10.3390/biom11050736
Zhang M, Yu Z, Zeng D, Si C, Zhao C, Wang H, Li C, He C, Duan J. Transcriptome and Metabolome Reveal Salt-Stress Responses of Leaf Tissues from Dendrobium officinale. Biomolecules. 2021; 11(5):736. https://doi.org/10.3390/biom11050736
Chicago/Turabian StyleZhang, Mingze, Zhenming Yu, Danqi Zeng, Can Si, Conghui Zhao, Haobin Wang, Chuanmao Li, Chunmei He, and Jun Duan. 2021. "Transcriptome and Metabolome Reveal Salt-Stress Responses of Leaf Tissues from Dendrobium officinale" Biomolecules 11, no. 5: 736. https://doi.org/10.3390/biom11050736
APA StyleZhang, M., Yu, Z., Zeng, D., Si, C., Zhao, C., Wang, H., Li, C., He, C., & Duan, J. (2021). Transcriptome and Metabolome Reveal Salt-Stress Responses of Leaf Tissues from Dendrobium officinale. Biomolecules, 11(5), 736. https://doi.org/10.3390/biom11050736