Abstract
Background: Amyotrophic Lateral Sclerosis (ALS) is a degenerative disorder which affects the motor neurons. Growing evidence suggests that ALS may impact the metabolic system, including the glucose metabolism. Several studies investigated the role of Diabetes Mellitus (DM) as risk and/or prognostic factor. However, a clear correlation between DM and ALS has not been defined. In this review, we focus on the role of DM in ALS, examining the different hypotheses on how perturbations of glucose metabolism may interact with the pathophysiology and the course of ALS. Methods: We undertook an independent PubMed literature search, using the following search terms: ((ALS) OR (Amyotrophic Lateral Sclerosis) OR (Motor Neuron Disease)) AND ((Diabetes) OR (Glucose Intolerance) OR (Hyperglycemia)). Review and original articles were considered. Results: DM appears not to affect ALS severity, progression, and survival. Contrasting data suggested a protective role of DM on the occurrence of ALS in elderly and an opposite effect in younger subjects. Conclusions: The actual clinical and pathophysiological correlation between DM and ALS is unclear. Large longitudinal prospective studies are needed. Achieving large sample sizes comparable to those of common complex diseases like DM is a challenge for a rare disease like ALS. Collaborative efforts could overcome this specific issue.
1. Introduction
Amyotrophic Lateral Sclerosis (ALS) is a degenerative disorder of unknown cause which affects the motor neurons of the cerebral cortex, brainstem, and spinal cord [1,2,3,4].
The primary triggers for motor neuron degeneration in ALS remain elusive, however research in patients and in SOD1 mutant mice models has revealed several processes that are likely to contribute to the pathology, including inflammation and toxic glial activation [5,6], mitochondrial dysfunction [7,8], and oxidative stress [9,10].
Despite the traditional view of ALS as a pure motor neuron disorder, growing evidence suggests that ALS may impact different systems, the including cellular metabolic system [11,12,13].
Energy homeostasis requires that uptake of nutrients in cells, including glucose and lipids, is adequately controlled by the glucose–insulin axis [11].
Several studies demonstrated that two-thirds of ALS patients develop a stable hypermetabolism during the course of the disease [11,14,15,16] which has been linked to a worse prognosis in several studies [17]. However, there is no consensus in literature and, although several hypothesis were examined, the mechanisms underlying hypermetabolism are still unclear [11,14,18,19,20].
Dupuis et al. observed a protective role of hyperlipidemia in rodents with ALS and showed how a high Low Density Lipoprotein (LDL)/High Density Lipoprotein (HDL) ratio in ALS patients, which is a measure of high levels of fat storage, correlates with a better prognosis, demonstrating a favorable prognostic role of hyperlipidemia in ALS patients. These data reflect evidence that high-calorie diets improve prognosis increasing the energy and consequently reducing the energetic deficit developed in motor neurons, avoiding the development of a hypermetabolic state which particularly involves lipid metabolism. In fact, to support the increased energetic demand of denervated muscle a higher amount of free fatty acids converted through lipolysis is required [20,21].
In line with these findings, it has been observed that malnutrition negatively affects ALS patients. Moreover, a low (under-weight) pre-morbid Body Mass Index (BM) and a faster weight loss after disease onset are linked to a worse prognosis [20,22,23,24,25,26]
Contradictory findings have been reported about glucose metabolism. Some authors described the occurrence of insulin resistance during the course of ALS [27,28,29,30] and several studies investigated the role of Diabetes Mellitus (DM) as risk and/or prognostic factor [17,26,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50].
However, it is still difficult to define a clear correlation between DM and ALS because results are conflicting and the possible underlying common pathophysiology is still elusive [13,31,44,47,48,51].
In this review, the focus is on the role of DM in ALS and different hypotheses are presented concerning how perturbations of glucose metabolism may interact with the pathophysiology and the course of the disease.
2. Materials and Methods
The authors undertook an independent PubMed literature search, using the following search formula: ((ALS) OR (Amyotrophic Lateral Sclerosis) OR (Motor Neuron Disease)) AND ((Diabetes) OR (Glucose Intolerance) OR (Hyperglycemia)).
Both review and original articles were considered. The preferred reporting items for systematic reviews and meta-analyses is given in a diagram describing the research from literature (Figure 1) [52].
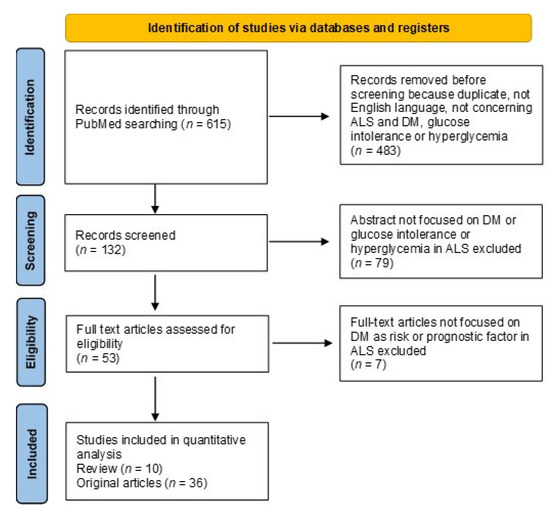
Figure 1.
The flow-chart depicts the different phases of the literature search and analysis.
3. Results
Results reported below in narrative description are also summarized in Table 1.

Table 1.
Summary of the correlation between main issue and ALS course and prognosis.
3.1. Diabetes Mellitus as a Risk Factor for ALS
Diabetes has been studied as a risk factor for ALS, with conflicting results.
The pioneer among the studies on DM in Caucasian ALS patients is the study by Jawaid et al. [33] which found that premorbid DM2 was associated with a reduced risk of ALS and a 4 years-delayed onset compared to a group without DM2 (ALS-DM = 60.3; ALS = 56.3; p < 0.05) [33].
According to Jawaid et al., some authors documented a lower prevalence of DM in ALS patients compared to a control cohort matched for age and gender and this difference has been read as an expression of lower susceptibility to ALS of diabetic patients [32,36,38,39,53]. Thus, they suggested DM as a protective factor against ALS occurrence [32,36,38,39].
One potential explanation might be a selection bias which led investigators to recruit patients without comorbidities in an ALS-related clinical trial [69]. Secondly, ALS trial participants were often defined as diabetic based on self-reported medical history or retrospectively using International Classification of Disease (ICD) codes which can underestimate the prevalence of DM [41,54].
Concerning the effects of DM on age at onset of ALS, some authors reported delayed ALS onset in DM patients [39,41,49,55], while other population-based studies did not find differences of age at onset between ALS patients with or without DM [40].
Two north-European case-control studies showed that the risk of ALS was higher among subjects with a diagnosis of diabetes before the age of 40, while it was lower in those with a diabetes onset after 40 years of age [36,38].
Two recent population-based studies, the former conducted in China [42] and the latter in Taiwan [56], reported results consistent with previous studies performed in Western countries [33,55,57].
In particular, Tsai et al. performed a case-control population-based study where they found that hypometabolic disorders, especially DM, have a beneficial effect on ALS incidence [58]. After that, the same group elaborated more deeply the data about DM in ALS and they found that the onset of ALS was delayed by 4 years in DM2 patients, highlighting a protective role of DM2 on ALS onset, especially in patients older than 55 years [56].
Other studies conducted in Asian populations showed different findings. Sun et al. reported a significantly increased risk of ALS in diabetic patients compared to non-diabetics with an overall hazard ratio (HR) of 1.35. After adjustment for age, they showed an increased HR in younger diabetic men (<65 years) while for older diabetic men and women (>65 years), the risk of ALS maintained the same trend but was not significant [43,44].
In conclusion it seems that in younger patients, diabetes is a consistent risk factor for ALS. In older patients, diabetes protects against ALS in Caucasians but increases the risk in Asian populations [13].
These findings may be explained by considering the substantial differences of ALS between European and Asian populations in terms of clinical features and potential molecular mechanisms, in terms of possible biological pathways underlying ALS in different populations [37] and of the genetic basis of ALS in the two populations [37,44].
3.2. Diabetes Mellitus as a Prognostic Factor for ALS
Several studies investigated the association among comorbidities and ALS survival and specifically between DM and ALS. All these studies similarly reported that DM2 did not affect the course of the disease in terms of survival or in terms of ALS Functional Rating Scale Revised (ALSFRSR) and respiratory function decline [40,41,45,49,50,59].
Conversely, an assessment of the correlation with baseline glycated hemoglobin (HbA1c) and survival in a Chinese ALS cohort of patients, classified into three groups based on their HbA1c levels (<5.7%, 5.7–6.4%, and >6.4%) using the group with the lowest HbA1c level as reference, showed that increased baseline HbA1c was associated with increased risk of mortality for any cause, after adjustment for epidemiologic and clinical covariates (i.e., age, sex, site of onset, rate of disease progression, BMI, use of riluzole, gastrostomy percutaneous endoscopy, noninvasive positive pressure ventilation) [60].
Nevertheless, loss/dysregulation of energy homeostasis has been proposed to negatively impact the course of the disease with a greater LMN involvement, worse functional decline over time and reduced survival [16,17].
De la Rubia Orti et al. performed a cross-sectional study in which they investigated the role of hyperglycemia and insulin resistance index, defined as the correlation of glucose venous blood value with increased Alkaline Phosphatase (AP). A significant positive correlation was observed between insulin resistance and spinal ALS onset [61].
Regarding cognitive performance it has been reported that pre-morbid DM was associated with either similar or worse cognitive performance on neuropsychological tests at the time of ALS diagnosis [46]. Specifically, ALS-DM performed worse on tests of memory, semantic fluency, and executive function [33].
It is unclear why diabetes would have a protective effect on the motor symptoms but not on the cognitive symptoms of ALS [12].
4. Discussion
Despite in literature the presence of perturbations in glucose metabolism in patients with ALS has been repeatedly reported, the question about how DM and ALS interact is still open.
It is unclear if DM is a manifestation of ALS rather than a protective factor [35]. To elucidate the molecular mechanisms underlying the extra-neural pathology of ALS, some authors focus on TAR DNA-binding protein 43 kDa (TDP-43) which is found in 90–95% of sporadic ALS patients, and that in familial ALS is linked to TAR DNA-binding protein (TARDBP) or C9orf72-SMCR8 Complex Subunit (C9ORF72) genes. TDP-43 pathophysiology is linked to the loss of nuclear protein and appearance of cytoplasmatic aggregations in motor neurons [70,71].
It is known that TDP-43 can regulate whole-body metabolism and glucose transport through the obesity-related gene TBC1 domain family member 1 (Tbc1d1) [12]. Recently, it has been observed that a loss of nuclear TDP-43 underlies the impaired early-phase secretion of insulin which is observed in early-stage ALS patients [12,72,73,74]. Araki et al. proposed a possible role for nuclear TDP-43 depletion in pancreatic β cells in the decreased insulin secretion of patients with sporadic ALS [73].
Moreover, it has been observed that SOD1 mice exhibit signs of metabolic dysfunction even at the pre-symptomatic stage of ALS, with dysregulation of lipid and carbohydrate metabolism [19,20].
An exon-array analysis showed that C9ORF72 repeat expansions lead to a differentially regulated splicing of several genes involved in cholesterol biosynthesis and glucose metabolism [12,75,76,77].
Furthermore, some authors speculated that the protective role of DM on ALS onset might be due to the effect of anti-diabetic drugs [62]. More specifically, two different studies investigated the role of pioglitazone and metformin in patients with ALS. Dupuis et al. reported a phase II, multicentric, placebo-controlled trial of pioglitazone in ALS patients under riluzole treatment regimen. Pioglitazone is used in clinical practice as an anti-diabetic agent; however, it also plays a role as an anti-oxidant and anti-inflammatory agent, which made it a candidate for ALS treatment since oxidative stress and inflammation are implicated in ALS pathophysiology [78]. Pre-clinical studies in SOD1 transgenic mice showed positive results [79,80,81].
Notwithstanding the promising pre-clinical data, Dupuis et al. had to stop that trial for futility, in absence of effects on survival and secondary outcomes as ALSFRS-R score and decline of respiratory functions [63].
Similarly, Kaneb et al. performed a pre-clinical study with metformin in SOD1 transgenic mice on the basis of its anti-inflammatory [82,83,84] and anti-oxidant [85,86] properties [87]. They observed a lack of efficacy on survival in male mice and a negative effect on onset and progression in female mice. Therefore they suggest that metformin is a poor candidate for clinical trials in ALS patients [64].
Concerning pioglitazone, it has been suggested that the lack of efficacy could be due to the interaction between beneficial molecular mechanism of the drug and this opposite detrimental effect on the whole-body metabolism, i.e., weight loss which is associated with a faster disease progression [57,65]. Moreover, some authors speculated that the unexpected failure of anti-diabetic drugs in ALS might be due to the nullification of the supposed beneficial effects of DM-associated hypermetabolism in ALS patients [65].
In fact, the third mechanism proposed is that high levels of glucose in ALS patients could reduce the damage caused to motor neurons by hypermetabolism.
Hypermetabolism is defined as a significant increase in measured resting energy expenditure (REE) relative to predicted REE, based on the Harris–Benedict equation [14,15,20,88] or measurements including fat-free mass [16].
Several studies suggested that in an attempt to avoid an increase in oxidative stress in the pre-symptomatic stage of the disease [11,15,39], mitochondria become uncoupled, ensuing in hypermetabolism [89,90]. Hence, higher resting metabolism could be activated to maintain energetic status [13].
This hypermetabolism may appear paradoxical because Free Fat Mass (FFM), the main determinant of REE, decreases in ALS [14,15,88]. Although many hypotheses have been proposed, such as increased respiratory muscular work [88], hypothalamic involvement [16], hyperthyroidism, spasticity or fasciculation intensity [18], hypermetabolism remains largely unexplained.
As above mentioned, several studies reported that hypermetabolic patients with ALS have a trend towards shorter survival [16,91]. Thus, hypermetabolism has been indicated to be a negative prognostic factor for ALS.
In this context, some authors speculated that DM positive effects should be the results of the activation of alternative pathways, such as ketogenesis, which are metabolically more efficient than glucose or fats in mitochondrial ATP generation [92,93,94].
Moreover, Jawaid et al. suggested that because the formation of stress granules might be induced by glucose deprivation, factors that lead to glucose starvation might enhance formation of TDP-43/FUS aggregation, and conversely DM2 should provide protection by supplying more glucose or alternate sources of energy preventing formation of stress granules [12]. Further, it has been proposed that DM could exert its protective role through hyperglycemia, which has been described to promote glutamate uptake [36,95].
Indeed, it has been demonstrated that a high-caloric diet, with either high carbohydrate or high fat contents, improves prognosis [20,24,66,96,97,98] increasing energy availability and consequently reducing the energy deficit developed in motor neurons [20].
Nevertheless, contrasting data, resulting from the analysis of glucose pathway, underline how abnormalities in the course of glucose metabolism pathway can lead to decreased generation of adenosine triphosphate (ATP) and a subsequent activation of compensatory alternative energy generating pathways which may result in increased oxidative stress [51,99,100,101].
Furthermore, some authors suggested that hypermetabolism may not be a direct factor influencing prognosis, but rather, a consequence of the rearrangement of whole-body metabolism and the activation of less efficient energy pathways due to enhanced request or absence of metabolic substrate during the course of the disease, often present due to malnutrition as a result of dysphagia, inability of feeding oneself, fear of choking, and aspiration pneumonia [14,22,91,102].
Fourth, it is possible that the deep differences in natural disease histories of DM and ALS may confound the reported low prevalence of DM in ALS. Indeed, it is notable that hyperlipidemia, which has been shown to have a protective role in ALS [21], is frequent in DM patients, while a slim body and athletic life-style, suggested as risk factors for ALS, are uncommon in DM patients [4,103,104]. Hence, DM patients could have a less malignant course of ALS because of either the presence of a protective factor (hyperlipidemia) or absence of certain risk factors (low BMI and active lifestyle) [33].
Finally, Apolipoprotein E (APoE) has been proposed to be involved in the pathophysiology of ALS and to influence the course of ALS. ApoE is the key regulator of plasma lipids, mediating altered functions in lipoprotein metabolism. ApoE genotype has been observed to be implicated as a risk factor in several neurological disorders such as Alzheimer’s Disease, Creutzfeldt-Jacob disease, Wilson’s disease, Multiple Sclerosis, cerebral amyloid angiopathy, Parkinson’s Disease and Lewy Body Dementia [105,106,107,108,109,110,111,112,113]. It has been speculated that its impact in neurological diseases is due to its role in lipid transport, neuronal repair, calcium homeostasis, and antioxidant activity [66,88,89,90,96].
Some studies observed a higher age of onset of ALS in patients carrying the ApoE2 allele [114,115], while a lower age of onset was detected in subjects with Apoe4 allele [116,117]. However, no correlation with the clinical course of ALS was detected in association with the ApoE genotype [67,118]. ApoE plasma levels have been correlated with a worse rate of physical deterioration and shorter survival time [118].
Both ApoE2 and ApoE4 have been found to be over-expressed in diabetic and hyperlipidemic populations in North America [119]. Consequently, some authors suggested that the supposed beneficial effects of DM on ALS are due to the ApoE genotype which may independently influence the risk of developing DM and the onset and progression of ALS [33].
5. Conclusions
According to our systematic review, DM appears not to affect disease severity, progression, and survival in ALS patients. On the other hand, contrasting data suggested a protective role of DM on the occurrence of ALS in elderly subjects and an inverse correlation in younger. But, there is still no consensus and the real clinical and pathophysiological correlation between DM and ALS is unclear. We describe how the multiple attempts to shed light on the relationship between DM and ALS may be hindered by several shortcomings.
The major limit of the above-mentioned studies is the difficulty in designing a study considering the dynamic course of DM, including measures of glycaemia control, and taking into account the wide range of co-variables associated with DM, as short and long term DM complications, in patients with ALS, which should partially justify the heterogeneity of currently available data.
On this basis we believe that it could be useful, in further studies, to assess an accurate distinction between DM1 and DM2 [68]. Indeed, many authors identified diagnosis of DM using International Classification of Disease (ICD) codes extrapolated from registers which enabled them to classify DM in type 1 and type 2 sometimes only based on insulin dependence [33,36,38,39]. This inaccurate distinction is relevant because available data suggest a different interaction between ALS and DM1 and DM2, explained by distinct pathophysiology of DM1 which recognize an autoimmune genesis, notably associated to an increase risk of ALS [120].
In this regard, Kioumourtzoglou et al. reanalysed previously collected data and re-classified part of their DM2 cohort as DM1, and found an overall protective association between DM2 and ALS diagnosis, while DM1 would represent a risk factor for ALS [36].
Moreover, Jawaid suggested that not only the pathophysiology should be considered but also the differences in body phenotype between DM1 and DM2. In particular, DM1 is more often associated with weight loss, which is associated with a faster ALS progression [33].
Despite discrepancies and limits of the available studies, considering the growing evidence of insulin-resistance involvement also in other neurodegenerative diseases such as Alzheimer’s Disease and Parkinson’s Disease [121,122,123,124], we suggest that further efforts in determining the role of glucidic metabolism in ALS would be crucial. Therefore, to obtain more reliable results, large longitudinal prospective studies are needed.
Notably, achieving large sample sizes comparable to those of common complex diseases like DM is a challenge for a rare disease like ALS. Collaborative efforts would hopefully overcome this specific challenge.
Author Contributions
Conceptualization, L.F., P.A., M.G.R. and L.B.; methodology, L.F., C.C., L.B.; writing—original draft preparation, L.F., P.A., M.G.R., C.C., F.B., D.D., M.C., A.D.M., F.C. and L.B.; writing—review and editing, L.F. and L.B.; visualization, L.B.; supervision, L.B., M.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; D’Ovidio, F.; Vasta, R.; Grassano, M.; Brunetti, M.; Barberis, M.; Corrado, L.; et al. ALS phenotype is influenced by age, sex, and genetics. Neurology 2020, 94, e802–e810. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R. CLINICAL EPIDEMIOLOGY OF AMYOTROPHIC LATERAL SCLEROSIS. Neurol. Clin. 1996, 14, 399–420. [Google Scholar] [CrossRef]
- Brooks, B.R. Natural history of ALS: Symptoms, strength, pulmonary function, and disability. Neurology 1996, 47, 71–82. [Google Scholar] [CrossRef]
- Papadimitriou, D.; Le Verche, V.; Jacquier, A.; Ikiz, B.; Przedborski, S.; Re, D.B. Inflammation in ALS and SMA: Sorting out the good from the evil. Neurobiol. Dis. 2010, 37, 493–502. [Google Scholar] [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef]
- Nutini, M.; Frazzini, V.; Marini, C.; Spalloni, A.; Sensi, S.L.; Longone, P. Zinc pre-treatment enhances NMDAR-mediated excitotoxicity in cultured cortical neurons from SOD1G93A mouse, a model of amyotrophic lateral sclerosis. Neuropharmacol. 2011, 60, 1200–1208. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Goodall, E.F.; Morrison, K.E. Amyotrophic lateral sclerosis (motor neuron disease): Proposed mechanisms and pathways to treatment. Expert Rev. Mol. Med. 2006, 8, 1–22. [Google Scholar] [CrossRef]
- Takei, K.; Watanabe, K.; Yuki, S.; Akimoto, M.; Sakata, T.; Palumbo, J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 5–10. [Google Scholar] [CrossRef]
- Dupuis, L.; Pradat, P.-F.; Ludolph, A.C.; Loeffler, J.-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Jawaid, A.; Khan, R.; Polymenidou, M.; Schulz, P.E. Disease-modifying effects of metabolic perturbations in ALS/FTLD. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef]
- Brito, M.D.; Da Silva, G.F.G.; Tilieri, E.M.; Araujo, B.G.; Calió, M.L.; Rosenstock, T.R. Metabolic Alteration and Amyotrophic Lateral Sclerosis Outcome: A Systematic Review. Front. Neurol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Desport, J.-C.; Torny, F.; Lacoste, M.; Preux, P.-M.; Couratier, P. Hypermetabolism in ALS: Correlations with Clinical and Paraclinical Parameters. Neurodegener. Dis. 2005, 2, 202–207. [Google Scholar] [CrossRef]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Ioannides, Z.A.; Van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; Berg, L.H.V.D.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S. Hypermetabolism appears to be an adverse prognostic biomarker in amyotrophic lateral sclerosis: A potential for therapeutic intervention? Eur. J. Neurol. 2017, 25, 1–2. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrère, B.; Couratier, P. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef]
- Fergani, A.; Oudart, H.; De Aguilar, J.-L.G.; Fricker, B.; René, F.; Hocquette, J.-F.; Meininger, V.; Dupuis, L.; Loeffler, J.-P. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J. Lipid Res. 2007, 48, 1571–1580. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; Rene, F.; de Aguilar, J.-L.G.; Loeffler, J.-P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; De Aguilar, J.-L.G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. On Behalf of the Eurals Consortium Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Truong, T.C.; Vallat, J.M.; Sautereau, D.; Couratier, P. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999, 53, 1059. [Google Scholar] [CrossRef] [PubMed]
- Pape, J.; Grose, J. The effects of diet and sex in amyotrophic lateral sclerosis. Rev. Neurol. 2020, 176, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Chełstowska, B.; Kuźma-Kozakiewicz, M. Biochemical parameters in determination of nutritional status in amyotrophic lateral sclerosis. Neurol. Sci. 2020, 41, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, A.; Murthy, S.B.; Wilson, A.M.; Qureshi, S.U.; Amro, M.J.; Wheaton, M.; Simpson, E.; Harati, Y.; Strutt, A.M.; York, M.K.; et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph. Lateral Scler. 2010, 11, 542–548. [Google Scholar] [CrossRef]
- Pradat, P.-F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.-M.; et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 166–171. [Google Scholar] [CrossRef]
- Harno, K.; Rissanen, A.; Palo, J. Glucose tolerance in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2009, 70, 451–455. [Google Scholar] [CrossRef]
- Reyes, E.T.; Perurena, O.H.; Festoff, B.W.; Jorgensen, R.; Moore, W.V. Insulin resistance in amyotrophic lateral sclerosis. J. Neurol. Sci. 1984, 63, 317–324. [Google Scholar] [CrossRef]
- Shimizu, T.; Honda, M.; Ohashi, T.; Tsujino, M.; Nagaoka, U.; Kawata, A.; Watabe, K.; Matsubara, S.; Hayashi, H. Hyperosmolar hyperglycemic state in advanced amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 12, 379–381. [Google Scholar] [CrossRef]
- Lekoubou, A.; Matsha, T.E.; Sobngwi, E.; Kengne, A.P. Effects of diabetes mellitus on amyotrophic lateral sclerosis: A systematic review. BMC Res. Notes 2014, 7, 171. [Google Scholar] [CrossRef]
- D’Ovidio, F.; D’Errico, A.; Carnà, P.; Calvo, A.; Costa, G.; Chiò, A. The role of pre-morbid diabetes on developing amyotrophic lateral sclerosis. Eur. J. Neurol. 2017, 25, 164–170. [Google Scholar] [CrossRef]
- Jawaid, A.; Salamone, A.R.; Strutt, A.M.; Murthy, S.B.; Wheaton, M.; McDowell, E.J.; Simpson, E.; Appel, S.H.; York, M.K.; Schulz, P.E. ALS disease onset may occur later in patients with pre-morbid diabetes mellitus. Eur. J. Neurol. 2010, 17, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, A.; Schulz, P.E. Pre-morbid type 2 diabetes mellitus as a prognostic factor in amyotrophic lateral sclerosis. Muscle Nerve 2015, 52, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, A.; Brown, J.A.; Schulz, P.E. Diabetes mellitus in amyotrophic lateral sclerosis: Dr. Jekyll or Mr. Hyde? Eur. J. Neurol. 2015, 22, 1419–1420. [Google Scholar] [CrossRef]
- Kioumourtzoglou, M.-A.; Rotem, R.S.; Seals, R.M.; Gredal, O.; Hansen, J.; Weisskopf, M.G. Diabetes Mellitus, Obesity, and Diagnosis of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2015, 72, 905–911. [Google Scholar] [CrossRef]
- Logroscino, G. Are diabetes and amyotrophic lateral sclerosis related? Nat. Rev. Neurol. 2015, 11, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Mariosa, D.; Kamel, F.; Bellocco, R.; Ye, W.; Fang, F. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur. J. Neurol. 2015, 22, 1436–1442. [Google Scholar] [CrossRef]
- Mariosa, D.; Kamel, F.; Bellocco, R.; Ronnevi, L.; Almqvist, C.; Larsson, H.; Ye, W.; Fang, F. Antidiabetics, statins and the risk of amyotrophic lateral sclerosis. Eur. J. Neurol. 2020, 27, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Moglia, C.; Calvo, A.; Canosa, A.; Bertuzzo, D.; Cugnasco, P.; Solero, L.; Grassano, M.; Bersano, E.; Cammarosano, S.; Manera, U.; et al. Influence of arterial hypertension, type 2 diabetes and cardiovascular risk factors on ALS outcome: A population-based study. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 590–597. [Google Scholar] [CrossRef]
- Paganoni, S.; Bs, T.H.; Shui, A.; Allred, P.; Harms, M.; Liu, J.; Maragakis, N.; Schoenfeld, D.; Yu, H.; Atassi, N.; et al. Pre-morbid type 2 diabetes mellitus is not a prognostic factor in amyotrophic lateral sclerosis. Muscle Nerve 2015, 52, 339–343. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, L.; Fan, D. The protective role of pre-morbid type 2 diabetes in patients with amyotrophic lateral sclerosis: A center-based survey in China. Amyotroph. Lateral Scler. Front. Degener. 2019, 21, 209–215. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, C.-J.; Chen, R.-C.; Hou, W.-H.; Li, C.-Y. Risk of Amyotrophic Lateral Sclerosis in Patients With Diabetes: A Nationwide Population-Based Cohort Study. J. Epidemiol. 2015, 25, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Wang, T.; Zheng, J.; Zhou, X. Causal association of type 2 diabetes with amyotrophic lateral sclerosis: New evidence from Mendelian randomization using GWAS summary statistics. BMC Med. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Ferri, L.; Fasano, A.; Zucchi, E.; Fini, N.; Moglia, C.; Lunetta, C.; Marinou, K.; Ticozzi, N.; Ferrante, G.D.; et al. Cardiovascular diseases may play a negative role in the prognosis of amyotrophic lateral sclerosis. Eur. J. Neurol. 2018, 25, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; ERRALS Group; Malerba, S.A.; Beghi, E.; Fini, N.; Fasano, A.; Zucchi, E.; De Pasqua, S.; Guidi, C.; Terlizzi, E.; et al. Riluzole and other prognostic factors in ALS: A population-based registry study in Italy. J. Neurol. 2018, 265, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Vasta, R.; D’Ovidio, F.; Logroscino, G.; Chiò, A. The links between diabetes mellitus and amyotrophic lateral sclerosis. Neurol. Sci. 2021, 42, 1377–1387. [Google Scholar] [CrossRef]
- Wannarong, T.; Ungprasert, P. Diabetes mellitus is associated with a lower risk of amyotrophic lateral sclerosis: A systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2020, 199, 106248. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Peter, R.S.; Nagel, G.; Rothenbacher, D.; Rosenbohm, A.; Ludolph, A.C.; Dorst, J. The ALS Registry Swabia Study Group Statins, diabetes mellitus and prognosis of amyotrophic lateral sclerosis: Data from 501 patients of a population-based registry in southwest Germany. Eur. J. Neurol. 2020, 27, 1405–1414. [Google Scholar] [CrossRef]
- Bellavia, A.; Dickerson, A.S.; Rotem, R.S.; Hansen, J.; Gredal, O.; Weisskopf, M.G. Joint and interactive effects between health comorbidities and environmental exposures in predicting amyotrophic lateral sclerosis. Int. J. Hyg. Environ. Health 2021, 231, 113655. [Google Scholar] [CrossRef]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- Mariosa, D.; Hammar, N.; Malmström, H.; Ingre, C.; Jungner, I.; Ye, W.; Fang, F.; Walldius, G. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: A more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 2017, 81, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.S.; Hollinger, S.K.; Goswami, S.D.; Polak, M.A.; Lee, R.H.; Glass, J.D. Antecedent Disease Is Less Prevalent in Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2015, 15, 109–113. [Google Scholar] [CrossRef]
- Hollinger, S.K.; Okosun, I.S.; Mitchell, C.S. Antecedent Disease and Amyotrophic Lateral Sclerosis: What Is Protecting Whom? Front. Neurol. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-P.; Lee, J.K.-W.; Lee, C.T.-C. Type II diabetes mellitus and the incidence of amyotrophic lateral sclerosis. J. Neurol. 2019, 266, 2233–2243. [Google Scholar] [CrossRef]
- Jawaid, A.; Abid, A.; Schulz, P.E. Diabetes mellitus and amyotrophic lateral sclerosis: Time to bridge the gap between the bench and the bedside. Eur. J. Neurol. 2017, 25, 3–4. [Google Scholar] [CrossRef]
- Tsai, C.-P.; Hu, C.; Lee, C.T.-C. Finding diseases associated with amyotrophic lateral sclerosis: A total population-based case–control study. Amyotroph. Lateral Scler. Front. Degener. 2018, 20, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Körner, S.; Kollewe, K.; Ilsemann, J.; Müller-Heine, A.; Dengler, R.; Krampfl, K.; Petri, S. Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur. J. Neurol. 2012, 20, 647–654. [Google Scholar] [CrossRef]
- Wei, Q.-Q.; Chen, Y.; Cao, B.; Ou, R.W.; Zhang, L.; Hou, Y.; Gao, X.; Shang, H. Blood hemoglobin A1c levels and amyotrophic lateral sclerosis survival. Mol. Neurodegener. 2017, 12, 69. [Google Scholar] [CrossRef]
- Ortí, J.D.L.R.; Armero, J.; Sanchis-Sanchis, C.; Sancho-Castillo, S.; Salazar, A.; Caplliure-Llopis, J.; Navarro-Illana, E.; Barrios, C.; Escribá-Alepuz, J.; Benlloch, M. Muscle Function Differences between Patients with Bulbar and Spinal Onset Amyotrophic Lateral Sclerosis. Does It Depend on Peripheral Glucose? J. Clin. Med. 2021, 10, 1582. [Google Scholar] [CrossRef]
- Pfeiffer, R.M.; Mayer, B.; Kuncl, R.W.; Check, D.P.; Cahoon, E.K.; Rivera, D.R.; Freedman, D.M. Identifying potential targets for prevention and treatment of amyotrophic lateral sclerosis based on a screen of medicare prescription drugs. Amyotroph. Lateral Scler. Front. Degener. 2019, 21, 235–245. [Google Scholar] [CrossRef]
- Dupuis, L.; Dengler, R.; Heneka, M.T.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; Steiner, F.; Lindauer, E.; Otto, M.; et al. A Randomized, Double Blind, Placebo-Controlled Trial of Pioglitazone in Combination with Riluzole in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e37885. [Google Scholar] [CrossRef] [PubMed]
- Kaneb, H.M.; Sharp, P.S.; Rahmani-Kondori, N.; Wells, D.J. Metformin Treatment Has No Beneficial Effect in a Dose-Response Survival Study in the SOD1G93A Mouse Model of ALS and Is Harmful in Female Mice. PLoS ONE 2011, 6, e24189. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, A.; Paganoni, S.; Hauser, C.; Schulz, P.E. Trials of antidiabetic drugs in amyotrophic lateral sclerosis: Proceed with caution? Neurodegener. Dis. 2013, 13, 205–208. [Google Scholar] [CrossRef]
- Ferri, A.; Coccurello, R. What is “Hyper” in the ALS Hypermetabolism? Mediat. Inflamm. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Jawaid, A.; Poon, M.; Strutt, A.M.; Rice, L.K.; McDowell, E.J.; Salamone, A.R.; Qureshi, S.U.; Simpson, E.; Appel, S.H.; York, M.K.; et al. Does apolipoprotein E genotype modify the clinical expression of ALS? Eur. J. Neurol. 2010, 18, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Kawada, T. Type 2 diabetes and amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, e9. [Google Scholar] [CrossRef]
- Chio, A.; Canosa, A.; Gallo, S.; Cammarosano, S.; Moglia, C.; Fuda, G.; Calvo, A.; Gabriele, M. For the PARALS group ALS clinical trials: Do enrolled patients accurately represent the ALS population? Neurology 2011, 77, 1432–1437. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 2010, 40, 404–414. [Google Scholar] [CrossRef]
- Stallings, N.R.; Puttaparthi, K.; Dowling, K.J.; Luther, C.M.; Burns, D.K.; Davis, K.; Elliott, J.L. TDP-43, an ALS Linked Protein, Regulates Fat Deposition and Glucose Homeostasis. PLoS ONE 2013, 8, e71793. [Google Scholar] [CrossRef]
- Araki, K.; Araki, A.; Honda, D.; Izumoto, T.; Hashizume, A.; Hijikata, Y.; Yamada, S.; Iguchi, Y.; Hara, A.; Ikumi, K.; et al. TDP-43 regulates early-phase insulin secretion via CaV1.2-mediated exocytosis in islets. J. Clin. Investig. 2019, 129, 3578–3593. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Park, K.S.; Kim, S.H.; Yu, J.; Zhao, K.; Yu, L.; Oh, K.W.; Lee, K.; Kim, J.; Chaggar, K.; et al. IgGs from patients with amyotrophic lateral sclerosis and diabetes target CaVα2δ1 subunits impairing islet cell function and survival. Proc. Natl. Acad. Sci. USA 2019, 116, 26816–26822. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J.; et al. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, T.; Ji, Y.J.; Johnson, K.; Liu, H.; Johnson, K.; Bailey, S.; Suk, Y.; Lu, Y.-N.; Liu, M.; et al. A C9orf72–CARM1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes Dev. 2018, 32, 1380–1397. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J. C9orf72-dependent lysosomal functions regulate epigenetic control of autophagy and lipid metabolism. Autophagy 2019, 15, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Thiazolidinediones as anti-inflammatory and anti-atherogenic agents. Diabetes Metab. Res. Rev. 2007, 24, 14–26. [Google Scholar] [CrossRef]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar] [CrossRef]
- Shibata, N.; Kawaguchi-Niida, M.; Yamamoto, T.; Toi, S.; Hirano, A.; Kobayashi, M. Effects of the PPARγ activator pioglitazone on p38 MAP kinase and IκBα in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology 2008, 28, 387–398. [Google Scholar] [CrossRef]
- Schütz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schürmann, B.; Zimmer, A.; Heneka, M.T. The Oral Antidiabetic Pioglitazone Protects from Neurodegeneration and Amyotrophic Lateral Sclerosis-Like Symptoms in Superoxide Dismutase-G93A Transgenic Mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Ghanim, H.; Mohanty, P.; Tripathy, C.; Hofmeyer, D.; Chaudhuri, A. Increased Plasma Concentration of Macrophage Migration Inhibitory Factor (MIF) and MIF mRNA in Mononuclear Cells in the Obese and the Suppressive Action of Metformin. J. Clin. Endocrinol. Metab. 2004, 89, 5043–5047. [Google Scholar] [CrossRef]
- Isoda, K.; Young, J.L.; Zirlik, A.; Macfarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schönbeck, U.; Libby, P. Metformin Inhibits Proinflammatory Responses and Nuclear Factor-κB in Human Vascular Wall Cells. Arter. Thromb. Vasc. Biol. 2006, 26, 611–617. [Google Scholar] [CrossRef]
- Lin, H.Z.; Yang, S.Q.; Chuckaree, C.; Kuhajda, F.; Ronnet, G.; Diehl, A.M. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat. Med. 2000, 6, 998–1003. [Google Scholar] [CrossRef]
- Ouslimani, N.; Peynet, J.; Bonnefont-Rousselot, D.; Thérond, P.; Legrand, A.; Beaudeux, J.-L. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism 2005, 54, 829–834. [Google Scholar] [CrossRef]
- Srividhya, S.; Ravichandran, M.; Anuradha, C. Metformin Attenuates Blood Lipid Peroxidation and Potentiates Antioxidant Defense in High Fructose-fed Rats. J. Biochem. Mol. Biol. Biophys. 2002, 6, 379–385. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-Activated Protein Kinase Reduces Hyperglycemia-Induced Mitochondrial Reactive Oxygen Species Production and Promotes Mitochondrial Biogenesis in Human Umbilical Vein Endothelial Cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, E.J.; Berryman, S.; Vanderleest, J.G.; Schneider, A.R.; McClain, C.J. Nutritional status of patients with amyotrophic lateral sclerosis: Relation to the proximity of death. Am. J. Clin. Nutr. 1996, 63, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Funalot, B.; Desport, J.-C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Mead, R.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Jésus, P.; Fayemendy, P.; Nicol, M.; Lautrette, G.; Sourisseau, H.; Preux, P.-M.; Desport, J.-C.; Marin, B.; Couratier, P. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2018, 25, 97–104. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Caplliure-Llopis, J.; Peralta-Chamba, T.; Carrera-Juliá, S.; Cuerda-Ballester, M.; Drehmer-Rieger, E.; López-Rodriguez, M.M.; Ortí, J.E.D.L.R. Therapeutic alternative of the ketogenic Mediterranean diet to improve mitochondrial activity in Amyotrophic Lateral Sclerosis (ALS): A Comprehensive Review. Food Sci. Nutr. 2020, 8, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Wills, A.-M. High-Fat and Ketogenic Diets in Amyotrophic Lateral Sclerosis. J. Child Neurol. 2013, 28, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Patel, A.B.; Gjedde, A.; Rothman, D.L.; Behar, K.; Shulman, R.G. Neuronal–Glial Glucose Oxidation and Glutamatergic–GABAergic Function. Br. J. Pharmacol. 2006, 26, 865–877. [Google Scholar] [CrossRef]
- Wills, A.-M.; Hubbard, J.; Macklin, E.A.; Glass, J.; Tandan, R.; Simpson, E.P.; Brooks, B.; Gelinas, D.; Mitsumoto, H.; Mozaffar, T.; et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 383, 2065–2072. [Google Scholar] [CrossRef]
- Zhao, Z.; Sui, Y.; Gao, W.; Cai, B.; Fan, D. Effects of diet on adenosine monophosphate-activated protein kinase activity and disease progression in an amyotrophic lateral sclerosis model. J. Int. Med. Res. 2015, 43, 67–79. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H.M. A high fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T/ C57BL/6J mice. Dis. Model. Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2017, 10, 611. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; De León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef]
- Chio, A.; Calvo, A.; Ilardi, A.; Cavallo, E.; Moglia, C.; Mutani, R.; Palmo, A.; Galletti, R.; Marinou, K.; Papetti, L.; et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology 2009, 73, 1681–1685. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G. PARALS study group Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Scarmeas, N.; Helzner, E.; Glymour, M.M.; Brandt, J.; Albert, M.; Blacker, D.; Stern, Y. APOE 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology 2008, 70, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Schiefermeier, M.; Kollegger, H.; Madl, C.; Polli, C.; Oder, W.; Kühn, H.-J.; Berr, F.; Ferenci, P. The impact of apolipoprotein E genotypes on age at onset of symptoms and phenotypic expression in Wilson’s disease. Brain 2000, 123, 585–590. [Google Scholar] [CrossRef]
- Parmenter, B.A.; Denney, D.R.; Lynch, S.G.; Middleton, L.S.; Harlan, L.M. Cognitive impairment in patients with multiple sclerosis: Association with the APOE gene and promoter polymorphisms. Mult. Scler. J. 2007, 13, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Van Everbroeck, B.; Croes, E.A.; Pals, P.; Dermaut, B.; Jansen, G.; van Duijn, C.M.; Cruts, M.; Van Broeckhoven, C.; Martin, J.-J.; Cras, P. Influence of the prion protein and the apolipoprotein E genotype on the Creutzfeldt–Jakob Disease phenotype. Neurosci. Lett. 2001, 313, 69–72. [Google Scholar] [CrossRef]
- Ariza, M. Influence of APOE polymorphism on cognitive and behavioural outcome in moderate and severe traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1191–1193. [Google Scholar] [CrossRef]
- Louko, A.-M.; Vilkki, J.; Niskakangas, T. ApoE genotype and cognition after subarachnoid haemorrhage: A longitudinal study. Acta Neurol. Scand. 2006, 114, 315–319. [Google Scholar] [CrossRef]
- Tian, J.; Shi, J.; Bailey, K.; Lendon, C.L.; Pickering-Brown, S.; Mann, D.M.A. Association between apolipoprotein E e4 allele and arteriosclerosis, cerebral amyloid angiopathy, and cerebral white matter damage in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 696–699. [Google Scholar] [CrossRef]
- Li, Y.J.; Hauser, M.A.; Scott, W.K.; Martin, E.R.; Booze, M.W.; Qin, X.J.; Walter, J.W.; Nance, M.A.; Hubble, J.P.; Koller, W.C.; et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 2004, 62, 2005–2009. [Google Scholar] [CrossRef]
- Moulard, B.; Sefiani, A.; Laamri, A.; Malafosse, A.; Camu, W. Apolipoprotein E genotyping in sporadic amyotrophic lateral sclerosis: Evidence for a major influence on the clinical presentation and prognosis. J. Neurol. Sci. 1996, 139, 34–37. [Google Scholar] [CrossRef]
- Li, Y.-J.; Pericak-Vance, M.A.; Haines, J.L.; Siddique, N.; McKenna-Yasek, D.; Hung, W.-Y.; Sapp, P.; Allen, C.I.; Chen, W.; Hosler, B.; et al. Apolipoprotein E is associated with age at onset of amyotrophic lateral sclerosis. Neurogenetics 2004, 5, 209–213. [Google Scholar] [CrossRef]
- Drory, V.E.; Birnbaum, M.; Korczyn, A.D.; Chapman, J. Association of APOE ε4 allele with survival in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 190, 17–20. [Google Scholar] [CrossRef]
- Zetterberg, H.; Jacobsson, J.; Rosengren, L.; Blennow, K.; Andersen, P.M. Association of APOE with age at onset of sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2008, 273, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Lacomblez, L.; Doppler, V.; Beucler, I.; Costes, G.; Salachas, F.; Raisonnier, A.; Le Forestier, N.; Pradat, P.-F.; Bruckert, E.; Meininger, V. APOE: A potential marker of disease progression in ALS. Neurology 2002, 58, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Eichner, J.E.; Dunn, S.T.; Perveen, G.; Thompson, D.M.; Stewart, K.E.; Stroehla, B.C. Apolipoprotein E Polymorphism and Cardiovascular Disease: A HuGE Review. Am. J. Epidemiol. 2002, 155, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune disease preceding amyotrophic lateral sclerosis: An epidemiologic study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef]
- Shieh, J.C.-C.; Huang, P.-T.; Lin, Y.-F. Alzheimer’s Disease and Diabetes: Insulin Signaling as the Bridge Linking Two Pathologies. Mol. Neurobiol. 2020, 57, 1966–1977. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).