Abstract
Aberrant protein folding underpins many neurodegenerative diseases as well as certain myopathies and cancers. Protein misfolding can be driven by the presence of distinctive prion and prion-like regions within certain proteins. These prion and prion-like regions have also been found to drive liquid-liquid phase separation. Liquid-liquid phase separation is thought to be an important physiological process, but one that is prone to malfunction. Thus, aberrant liquid-to-solid phase transitions may drive protein aggregation and fibrillization, which could give rise to pathological inclusions. Here, we review prions and prion-like proteins, their roles in phase separation and disease, as well as potential therapeutic approaches to counter aberrant phase transitions.
1. Introduction
Protein folding is essential for life, as proteins serve structural roles, catalyze enzymatic reactions, and transport materials through membranes and cells, among many other functions [1]. Most globular proteins adopt a complex three-dimensional folded structure that is well-defined and associated with specific functions. In contrast, intrinsically disordered proteins (IDPs) do not adopt well-defined structures. Rather, IDPs occupy a highly dynamic ensemble of conformations, and these dynamic structural changes are key to the function of these proteins [2]. While some proteins are fully disordered in their native states, although they may adopt a folded state under certain conditions such as upon interaction with a binding partner, numerous proteins contain both well-folded regions and intrinsically disordered regions (IDRs) [3]. Up to 44% of known proteins are thought to contain at least one IDR of 30 residues or longer [4]. While globular proteins are driven to fold by the propensity of hydrophobic amino acids to pack in the core, disordered sequences have fewer hydrophobic residues, and as such are not driven to collapse by hydrophobic packing [5,6]. Rather, disordered proteins are enriched in hydrophilic and charged residues which promote interactions with aqueous solvent, leading to extended conformations [5,7]. However, disordered proteins or regions can also occupy compact conformations depending on the amino acid sequence and environment [8]. Some intrinsically disordered proteins are enriched in a small subset of amino acids with specific characteristics, which are defined as low complexity domains (LCDs). Examples of LCDs include polyglutamine expansions and the Phe-Gly-rich repeats of nucleoporins [9,10]. With their lower stability and access to more conformations than globular proteins, IDPs, IDRs, and LCDs are also prone to misfolding and aggregation. Indeed, misfolding of such proteins is often associated with diseases such as certain myopathies and cancers, as well as numerous neurodegenerative diseases (NDs) including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD) [11,12].
Another type of neurological disorder that arises from protein misfolding are prion disorders, such as Creutzfeldt-Jakob disease (CJD) [13]. Prions are infectious proteins that can convert to an alternative conformation [14]. This alternative conformation typically harbors a region that adopts the amyloid fold. Amyloid is comprised of stacked beta sheets which can template the conversion of native protein to the amyloid fold [15]. In addition to driving disease, prions can also promote long-term memory formation in mammals and confer adaptive stress responses in yeast [16,17]. Many DNA- and RNA-binding proteins (RBPs) have prion-like domains (PrLDs) defined by their similar amino acid compositions to yeast prions [18]. Many of these RBPs, such as TDP-43, FUS, and hnRNPA1, also have IDRs or LCDs and have been implicated in NDs such as ALS and FTD [19]. Other amyloidogenic proteins such as amyloid-β (Aβ), tau, and α-synuclein (α-Syn) have some similarities with prions and drive several NDs such as AD and PD [20].
A striking feature of RBPs is their ability to drive liquid-liquid phase separation (LLPS) which is the formation of biomolecular condensates often enriched in RBPs and RNA [21,22]. This LLPS process is driven by multivalent interactions among biomolecules, such as RNA-protein binding and transient interactions between disordered protein sequences [21,22]. Many PrLD-containing RBPs have been found to function as scaffold proteins, meaning they are essential for the droplet structure [23]. PrLDs are suggested to have evolved to promote the recruitment of these problematic RBPs to condensates to preserve their solubility [24]. LLPS droplets frequently form and dissolve in response to cellular signals. However, droplets which do not dissolve properly can undergo an aberrant liquid-to-solid phase transition that leads to protein aggregation and fibrillization [25,26]. Chaperone proteins, which promote cellular proteostasis by regulating protein folding and degradation, can modulate droplet dynamics [27,28,29]. It has been proposed that sequestration of aggregation-prone proteins in liquid-like condensates could be protective [30,31] while an aberrant phase transition could lead to misfolding and formation of pathological inclusions [26]. As such, methods to counter aberrant phase transitions are under development as potential therapeutic targets for several NDs. Current avenues of exploration include the development of nucleic acids, small molecules, and chaperone proteins to modulate the material properties of LLPS condensates [30,32,33]. Here, we review prions and prion-like proteins, their roles in LLPS and disease, as well as possible therapeutic approaches to counter aberrant phase transitions.
2. Prions, Amyloid, and Prion-like Domains
Prions, or proteinaceous infectious particles, are a unique class of proteins [14]. A salient feature of prions is their infectious nature, characterized as their ability to spread or propagate between cells and organisms [24,34]. Prion proteins adopt a native, soluble conformation typically associated with a cellular function [14,34]. However, these proteins can convert into a characteristic highly stable prion state. In this prion state, the fold converts to the amyloid conformation, which typically is comprised of cross-beta strands [35]. Here, monomers adopt a beta sheet fold and stack with other monomeric units to form a long unbranched fibril [15]. The amyloid fold is a very stable protein conformation, and as such, amyloid is generally resistant to detergents, heat denaturation, and protease cleavage [36]. Conversion of proteins to the amyloid fold is typically driven by an amyloidogenic region within the protein, which is necessary and sufficient for fibrillization [37,38,39,40]. Conversion to the amyloid fold is thought to be irreversible. Once a small amount of protein accesses the amyloid fold, this small quantity of protein can serve as a template, nucleating and accelerating the conversion of remaining monomeric protein to the amyloid fold [24,34]. Prions are defined as infectious amyloids, but not all amyloidogenic proteins are classified as prions. Prion proteins can serve functional roles, but also cause several devastating disorders which typically affect the central nervous system. A salient feature of prions is their infectious nature, characterized as their ability to spread or propagate between cells and organisms.
2.1. Prions and Disease
Prion disorders are unique due to the nature of their transmissibility between organisms [41,42]. Prions were first recognized as nucleic acid free infectious particles in transmissible spongiform encephalopathy (TSE). TSEs are underpinned by a conformation change of human cellular prion protein (PrPC) to its pathological state PrP-scrapie (PrPSc) [14]. Prion seeds can spontaneously form from alterations in protein conformation, perhaps just due to ordinary protein conformational dynamics. Conversion to the prion form can also be induced by external stimuli, such as inoculation of a seed [43,44,45,46,47,48]. Different conformations of prions can give rise to distinct prion strains which can lead to varying phenotypes of disease, largely due to differences in neuronal vulnerability [49,50]. For example, as scrapie progresses in goats, most of the animals show an affected nervous system and a subset of goats also display an early scratching phenotype. When goats are inoculated with scrapie brain material from diseased goats, those that received material from goats displaying the scratching phenotype appeared to inherit this scratching phenotype. Conversely, goats inoculated from animals with the disease but without a scratching phenotype became ill, but did not inherit the scratching phenotype [49]. These results indicate that the two phenotypes are due to different strains of the scrapie prion [49]. Various strains of PrPSc give rise to TSEs in humans including sporadic and familial CJD, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker disease [13,34]. While CJD can be sporadic, familial forms of TSEs are associated with specific mutations in the PrP gene that accelerate its conversion to the prion conformation.
2.2. Amyloidogenic Proteins in Neurodegenerative Disorders
Several amyloidogenic proteins have similarities to prions and have been implicated in ND [20]. These include Aβ and tau in AD, polyglutamine expanded huntingtin in HD, and α-Syn in PD [51,52,53,54,55,56,57]. Like prions, these proteins adopt a cross-β fold upon aggregation and are extremely resilient to denaturation. These proteins can also seed their own fibrillization. Here, preformed amyloid fibrils, obtained from aggregate-containing lysate or synthetically formed fibrils, greatly enhance fibrillization. This seeding has been demonstrated to accelerate fibrillization of monomeric recombinant protein in cultured cells and in living animals [20,58,59,60]. As with prions, just a small quantity of amyloid-containing seed is required to robustly initiate amyloid formation [61]. Similar to studies employing PrP strains, it has been demonstrated that both tau and α-Syn can populate distinct conformations and that these strains can be perpetuated and are heritable in cultured cells [62,63]. Furthermore, while inoculations of preformed fibrils in animals initially result in localized pathology, spreading of pathology to distant regions of the brain has been observed in a time-dependent manner [60,64,65,66,67]. For example, localized injection of α-Syn fibrils in mice olfactory bulbs induced α-Syn pathology throughout the brain [68,69,70]. This evidence suggests that Aβ, tau, and α-Syn aggregates can undergo cell-to-cell transmission as the disease progresses. Further evidence of the possibilities for cell-to-cell transmission comes from therapeutic strategies wherein fetal dopaminergic neurons grafted in a PD patient were invaded with tau and α-Syn pathology at the time of autopsy [71]. Disorders such as AD, PD, and HD remain distinct from prion disorders because, while there is evidence of cell-to-cell transmission, there have been no identified cases of disease transmission between people [20,72,73,74,75].
2.3. Prions and Amyloids Can Serve Functional Roles
Functional prions and amyloid have been found to serve various roles in humans, as well as in bacteria and in yeast. Examples of functional amyloids in humans include premelanosome protein (PMEL) [76,77,78], peptide hormones [79], cytoplasmic polyadenylation element binding protein 3 (CPEB3) [80,81,82,83], and semenogelins [84,85,86]. PMEL fibrils form exclusively in melanosomes to promote melanin biosynthesis and stabilize organelle structure [76,77]. Cleavage of PMEL to produce amyloidogenic fragments is tightly regulated to only occur in melanosomes, preventing accumulation of PMEL throughout the cell [78,87]. PMEL fibrils catalyze melanin biosynthesis, concentrate melanin, and aid in melanin transport [78]. The conversion of CPEB3 from a soluble translational repressor to an amyloid translational activator is important in long-term memory facilitation in neurological synapses [81,88,89]. SUMOylation of soluble CPEB3 prevents aggregation of its N-terminal glutamine-rich LCD. Neuronal stimulation promotes deSUMOylation and ubiquitination of CPEB3, allowing closely regulated amyloid fibrillization [80,82].
In bacteria, amyloid is a key component of biofilms, which are highly stable heterogeneous matrices [90]. These extracellular structures consist of lipids, polysaccharides, and extracellular DNA in addition to amyloid, and provide scaffolds in which communities of bacteria can reside [91]. Upon taking up residence in these biofilm communities, bacteria can share resources and confer other beneficial traits, which enables rapid adaptation of the community to stressors. Ultimately, biofilms allow bacteria to thrive in harsh environments and acquire resistance to antibiotics [90,92,93].
Much of our understanding of prion biology comes from work in Baker’s yeast, Saccharomyces cerevisiae [94]. Yeast have harnessed prions as non-genetic elements of inheritance to allow for rapid adaptation to cellular stress [95,96]. In yeast, the prions [PSI+] and [URE3] are associated with traits that are heritable in a non-Mendelian fashion. These traits are conferred upon structural changes, whereby the endogenous proteins Sup35 and Ure2 adopt the amyloid fold [17,97,98]. Sup35, a translation termination factor, can convert to its prion conformation under stress as an adaptive response [17,99]. On conversion to the amyloid fold, Sup35 no longer serves to terminate translation allowing for stop codon read through, thereby promoting expression of cryptic genetic variations in 3′ untranslated regions. This is thought to allow for rapid adaptation, and therefore a growth advantage under certain conditions [16,95,100,101]. The [URE3] prion is associated with nitrogen catabolite repression, thereby allowing yeast to utilize diverse nitrogen sources. However, it has also been suggested that fungal prions can be detrimental to yeast. For instance, certain variants of [PSI+] and [URE3] confer growth defects [102,103]. In regulating prions, yeast employ a native prion disaggregase, Hsp104, which is a hexameric AAA+ ATPase that regulates prion formation, elimination, and propagation [104,105,106,107,108,109].
2.4. Prion-Like Domains in Human Proteins
The identification of multiple yeast prions has generated interest in better understanding the full complement of prion proteins. Biochemical screens were performed to identify yeast proteins with prion properties. These screens identified 24 new proteins with similar properties to [PSI+] and [URE3], such as the ability to self-assemble and the heritability of traits [110]. Additionally, the newly identified yeast prions were found to share similar amino acid compositions in their amyloidogenic regions, suggesting an identifiable characteristic of prion proteins. Based on these amino acid biases identified in yeast prions, the PLAAC algorithm was developed to search the human proteome for potential prions [111]. 246 candidate proteins were identified with regions of similar amino acid compositions to those of yeast prions. These domains were termed prion-like domains (PrLDs), accounting for ~1.2% of the nearly 20,000 genes screened [18,111,112,113,114]. Using gene ontology analysis, over half of the proteins identified in this study were predicted to bind DNA or RNA [18,114,115]. Strikingly, following their initial identification as putative prion-like proteins, many of these RBPs were later linked to neurodegenerative diseases such as ALS and FTD [18]. Some of the highest ranked PrLD-containing proteins—such as TDP-43, FUS, TAF15, EWSR1, hnRNPA1, hnRNPA2B1, and ATXN2—are known to form pathological inclusions in ALS and FTD patients [116]. TDP-43 aggregates are also observed in AD, HD, and PD patients [117]. The TDP-43 PrLD is essential for many of its native functions, such as mediating protein-protein interactions, recruitment to stress granules (SGs), and several functions such as miRNA biogenesis and splicing [118,119,120,121,122]. Mutations in TDP-43 and FUS have also been linked to ALS and FTD. Most TDP-43 pathogenic mutations are found in its PrLD and some of these mutations, including Q331K and M337V, have been shown to accelerate its aggregation [123,124]. RNA binding and the PrLD are required for TDP-43 toxicity in model organisms, indicating that both are necessary for misfolding and pathogenesis [125,126,127,128].
Human PrLD-containing proteins display many features similar to those of prions. For instance, these PrL proteins can seed aggregation, form unique strains, induce neurotoxicity, and, in some cases, form amyloid-like fibrils [124,129,130,131,132]. PrLD-containing proteins are presumed to be noninfectious, but examples of cell-to-cell spreading throughout the brain have been observed [133]. Highly saturated concentrations of FUS, TDP-43, and SOD1 in spinal motor neurons suggest that these systems could be poised for LLPS and possibly prion-like propagation [134,135]. Recent studies suggest that while LLPS may be fundamental to TDP-43 function, malfunctioning of TDP-43 LLPS may lead to disease [29,30]. Neurodegenerative diseases typically develop later in life as proteostasis regulation progressively declines, leaving long-lived neurons less able to counter misfolding [136]. Many ALS cases are sporadic, and it is often unclear which mutations are causative. It is possible that conversion to a prion-like species triggers the onset of ALS pathogenesis [137].
3. Liquid-Liquid Phase Separation
In early studies investigating changes in nucleoli during the cell cycle, proteins in the nucleolus were found to have unusual properties. Various nucleoli-resident proteins were discovered to form dynamic droplets that could fuse on contact and exchange contents with materials in surrounding regions [138,139,140,141,142,143,144,145,146]. These droplet structures are now classified as membraneless organelles, also known as biomolecular condensates or LLPS droplets. LLPS droplets are formed by the demixing of associative biopolymers upon reaching their threshold concentration [21,22]. This demixing process yields high density droplets that display dynamic liquid-like properties including the capacity to undergo rapid internal rearrangements, exchange with surroundings, coalesce with other compatible droplets, and rebound to spherical shapes upon deformation [26]. Multivalent interactions drive this process, and can include specific RNA-protein interactions, specific protein-protein interactions, and nonspecific weak interactions between protein IDRs or LCDs [21,22]. As such, IDRs and PrLDs are often responsible for driving this demixing process [147,148]. Droplet formation is driven by scaffold proteins and many client proteins can be recruited to these droplets [149,150,151,152,153]. LLPS is believed to be an important physiological phenomenon that can drive essential processes, primarily by increasing the local concentration of specific molecules. For instance, certain biochemical reactions are thought to proceed only upon phase separation of necessary components, and corresponding exclusion of inhibitory particles [21,22]. LLPS can also be a protective process, such as the formation of SGs. For instance, the protein G3BP1 can function as a molecular switch that triggers LLPS in response to free RNA concentrations [154,155].
3.1. Role of Prion-Like Domains in LLPS
Many proteins with PrLDs have been found to drive or be recruited to LLPS droplets [23]. PrLDs can interact through weak nonspecific interactions, such as dipole-dipole interactions between polar residues and π-π interactions between aromatic residues [24]. Multivalent binding of RBPs to RNA may also contribute to this LLPS. As many of these PrLD-containing proteins serve essential roles in RNA binding, their condensation in LLPS droplets could allow for preservation of their solubility [151,156]. To enhance the solubility of aggregation-prone RBPs, PrLDs may have evolved to promote controlled condensate formation [24,157]. In support of this idea, RBPs with PrLDs such as Xvelo, RBM14, and hnRNPs have been recognized as scaffold proteins in various physiological membraneless organelles [23,158,159,160,161]. Recent studies have shown that amyloidogenic proteins such as tau and α-Syn, as well as the prions PrP and Sup35, also undergo LLPS [162,163,164,165]. Furthermore, condensate formation by PrP, tau, and α-Syn has been shown to trigger nucleation and fibrillization, suggesting that LLPS of these proteins may be directly linked to disease [162,164,165]. Such findings suggest that LLPS is a common mechanism that promotes aggregation of prions, amyloidogenic proteins, and PrLD-containing proteins.
3.2. Aberrant Phase Transitions and Protein Misfolding
In their physiological roles, it is thought that certain condensates frequently form and dissolve in response to external stimuli. However, under certain conditions, such as with aging, mutation, or chronic stress, LLPS droplets may not properly dissipate. When this occurs, the likelihood for misfolding and fibrillization is increased due to the high concentration of aggregation-prone proteins in condensates [25,26,166,167,168]. This can lead to aberrant solidification of the droplet as weak transient interactions within the droplet become more stable (Figure 1) [26]. These resulting highly cross-linked structures can be described as gel- or solid-like [21,22]. In contrast to their liquid-like precursors, gel- and solid-like structures do not display rapid internal rearrangements, do not exchange materials with their surroundings, and cannot readily dissipate [21,22]. Gel-like LLPS structures can also have physiological relevance, as yeast SGs are classified as more gel-like than liquid-like [169]. Some evidence indicates that aberrant LLPS could underpin neurodegenerative disease. Here, certain proteins may phase separate and dissipate repeatedly over time. Yet with aging or other stressors, these liquid-like condensates become more gel and solid-like, ultimately leading to the formation of the solid pathological inclusions observed in patients with neurodegenerative disease [25,26]. Such solid-like structures have been observed experimentally in droplet aging experiments [26].
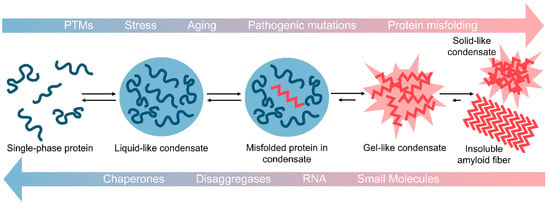
Figure 1.
Aberrant phase transitions drive protein fibrillization and aggregation. Many PrLD-containing proteins form liquid-liquid phase separated droplets comprised of protein and often RNA. These droplets have higher protein concentrations as compared to surrounding solutions, which can drive protein misfolding and aggregation. With aging, stress, post-translational modifications (PTMs), and/or pathogenic mutations, these liquid-like condensates can begin to solidify and form gel-like condensates. This process can progress further to produce solid-like condensates of aggregated protein or insoluble amyloid fibrils. Reversal of this process is of great therapeutic interest for many neurodegenerative diseases. Possible approaches to reverse aberrant phase transitions include application of chaperone proteins, protein disaggregases, specific RNA molecules, and small molecules.
Droplet dynamics can also be affected in other ways. For example, pathogenic mutations that destabilize proteins can accelerate droplet aging, which has been observed for several pathogenic mutations in the PrLD of TDP-43 [123,124,170]. Also, pathogenic mutations can increase mislocalization of RBPs to RNP granules, causing longer persistence of droplets [113,114,156,171,172]. Additionally, the partitioning of prion-like proteins to LLPS droplets can be modulated by interactions with binding partners and PTMs. For instance, binding of the nuclear import factor Karyopherin-β2 to FUS or methylation of FUS arginine residues both decrease its partitioning into SGs [28,32,173].
An independent disease pathway in ALS also results in impaired LLPS dynamics, emphasizing the importance of maintaining LLPS homeostasis. The most common genetic risk factor for ALS is a hexanucleotide repeat, G4C2, expansion in C9orf72 [174,175]. The presence of this expansion results in accumulation of toxic RNA species, production of dipeptide repeat proteins (DPRs) through repeat-associated non-AUG translation, and impairment of nucleocytoplasmic transport [176,177,178,179,180,181,182]. DPRs enriched in arginine (poly-glycine-arginine and poly-proline-arginine) can impair droplet dynamics [174,175,179,181]. These dipeptides can cause spontaneous assembly of SGs and decrease the saturation concentration of hnRNPA1 for droplet formation [181,182]. Arginine-rich DPRs can also trigger aggregation of TDP-43 [183,184]. Thus, multiple ALS pathways are converging on the disruption of condensate formation and dynamics, underscoring the critical importance of maintaining proper LLPS dynamics in neuronal health. Because of the close association between phase separation and proteins that underpin neurodegenerative disorders, there is intense interest in developing therapeutic modulators that restore physiological phase separation.
4. Chaperoning Phase Separation
Aberrant phase transitions are believed to underpin protein misfolding and disease; therefore, methods to counter aberrant phase transitions are an emerging area of research which holds great promise. A key goal is to preserve condensates in a liquid-like state and, perhaps more importantly, reverse the conversion to aberrant gel- and solid-like states. To accomplish this goal of preserving aggregation-prone proteins in liquid-like droplets and preventing conversion to pathological states, a range of approaches are being explored, including the development of chaperone proteins, RNA molecules, and small molecule inhibitors.
4.1. Protein Chaperones to Counter Aberrant Phase Transitions
Chaperones are a class of proteins that maintain cellular proteostasis by promoting proper protein folding and degradation, as well as preventing and countering protein aggregation [185]. RNP granules and SGs are comprised of several proteins and RNA. Interestingly, these structures are particularly enriched in chaperones such as Hsp40/70, which are likely important for droplet dissolution and maintenance of droplet dynamics [151,186,187,188,189,190]. For example, LLPS droplets of TDP-43 have been found to sequester Hsp70 within the droplet [29]. Hsp70 is also involved in preservation of phase separation in the nucleolus. Here, Hsp70 is required for recovery from heat stress, where it functions to extract and refold misfolded proteins [191]. Impairment of chaperone proteins or limited availability of ATP may hasten droplet aging and solidification [144,151,186].
Certain chaperone proteins have been found to not only prevent, but also reverse aberrant phase transitions. Protein disaggregases are a class of chaperone proteins that can both prevent and reverse protein aggregation. Hsp104 is an AAA+ native yeast protein disaggregase which regulates the assembly and disassembly of prions and also counters protein misfolding especially under stress [104,105,192,193,194,195,196,197]. In yeast SGs and P bodies, Hsp104 maintains droplet dynamics and regulates their dissolution [169]. Although metazoans lack an Hsp104 homolog, it was hypothesized that Hsp104 might be active against proteins that aggregate in human disease due to the conserved fold of amyloid and prion-like proteins. While activity of Hsp104 against many human disease-associated proteins is weak, potentiated variants of Hsp104 have been engineered that can prevent and reverse the misfolding of diverse proteins including TDP-43, FUS, and α-Syn [198,199,200,201,202,203,204,205,206,207,208,209,210,211]. Potentiated Hsp104 variants also displayed therapeutic activity in worm and mammalian cell models of neurodegenerative disease [198,212,213]. Other protein disaggregases have been identified, including Hsp110, Hsp70, Hsp40, as well as HtrA1, NMNAT2/Hsp90, TRIM11, and Karyopherin-β proteins [32,189,214,215,216,217,218]. Nuclear import receptors promote the transport of large proteins across the nuclear pore complex. Karyopherin-β2 (Kap-β2) is a nuclear import receptor that binds to and prevents the aggregation of RBPs including FUS, TAF15, EWSR1, hnRNPA1, and hnRNPA2, many of which accumulate in the cytoplasm in ALS and related disorders [32]. Kap-β2 can also dissolve fibrillized gel-like FUS condensates [32]. Additionally, Importin-α together with Karyopherin-β1 can counter and prevent fibrillization of TDP-43 [32]. It will be important to assess the activity of disaggregases and chaperones to reverse protein misfolding in solidified droplets. Ultimately, small-molecule therapeutics might be designed which stimulate disaggregase activity or promote the interaction of aggregation-prone proteins with disaggregases.
4.2. Applying RNA to Modulate Droplet Dynamics
RNA is a common component in biomolecular condensates due to its intrinsically multivalent properties. For instance, paraspeckles and P bodies are membraneless organelles which rely on RNA as a scaffold [219]. More broadly, RNA can have a variety of effects on droplet formation and dynamics. RNA can decrease the threshold concentration for phase separation driven by IDRs, which is the mechanism by which RNA modulates phase separation of hnRNPA1 in SGs and the phase separation of PGL-3 in P granules [25,168,220,221]. In contrast, in some systems, high concentrations of RNA can inhibit phase separation [222]. For example, phase separation of TDP-43 and FUS is inhibited in the RNA-rich nucleus but favored in the cytoplasm where RNA concentrations are lower [222]. Ultimately, this increased phase separation in the cytoplasm can lead to impairment of nuclear import. RNA composition and length have been found to correlate with droplet dynamics [166,223]. For instance, studies on phase separation of the polyglutamine-containing RBP, Whi3, demonstrated that short RNA molecules can decrease droplet dynamics while long RNA molecules have the opposite effect, likely due to RNA entanglement [22,166,223].
RNA molecules are also being explored as a method to alter the formation and dynamics of droplets for prevention of pathological aggregation. As proof of concept, Mann et al. demonstrated that bait RNA molecules binding TDP-43 can prevent aberrant phase transitions and subsequent neurotoxicity [30]. The same affect was not observed with a scrambled RNA control [30]. Additionally, they found that cytoplasmic inclusions of TDP-43 lack mRNA, suggesting that RNA may play a protective role by maintaining dynamic SGs and preventing TDP-43 aggregation [30]. In the future, tailored RNA molecules could be designed against specific therapeutic targets. Additionally, the development of other nucleic acid therapeutics such as antisense oligonucleotides have paved the way for RNA treatments.
4.3. Chaperoning Phase Transitions with Small Molecules
Condensates and their associated aggregation-prone proteins present challenges to traditional drug development approaches employing small molecules. These dynamic proteins often lack druggable binding pockets which small molecule therapeutics typically target [224]. As our understanding of phase separation and its corruption has grown, so has our understanding of how small molecules influence condensates, though several key questions remain [225]. To exploit small molecule therapeutics against condensates, it is important to understand how small molecules partition into droplets and how small molecules modulate the stability, dynamics, formation, composition, and other physical properties of condensates. Some small molecules such as 1,6-hexanediol broadly dissolve droplets of varied composition, but such molecules would be expected to disrupt many cellular processes reliant on LLPS [169,226]. Despite the highly dynamic nature of IDPs, small molecules can stabilize substrate-free, monomeric, or multimeric states of IDPs to alter their interactions, which are likely to influence condensate formation and properties [227,228,229]. For example, low concentrations of bis-ANS were found to initiate droplet formation of several proteins, including TDP-43, FUS, and tau, while dissolving droplets at high concentrations [33]. When a small molecule displays phase-dependent interactions with a protein, alterations to the phase diagram can be observed upon addition of the small molecule [230,231,232]. Small molecules can be designed to stabilize or destabilize LLPS assemblies by increasing or decreasing their multivalency, respectively [232]. Small molecules can also alter protein expression levels, such as Synucleozid which decreases α-Syn levels by targeting a regulatory element [233]. Beyond the direct decrease in α-Syn expression levels, such a strategy is also likely to alter condensate formation and offers another mechanism by which to target disordered proteins. Alternatively, a more traditional approach can use small molecule enzyme inhibitors to alter PTMs which govern LLPS droplet formation, composition, and dynamics [234]. Attachment of poly (ADP-ribose) (PAR), a negatively charged biopolymer, to target proteins controls localization and phase separation of targets such as TDP-43 and FUS [31,235]. Inhibiting PAR polymerases will downregulate this modification and limit its function in nucleating TDP-43 and FUS droplet formation [26,31,235,236].
Early studies aimed at drugging condensates with small molecules have proven promising. Studies by Klein et al. demonstrated that anti-cancer drugs such as cisplatin, mitoxantrone, and tamoxifen partition preferentially in nuclear transcription condensates. The drugs were recruited by aromatic and polar properties of the condensates, rather than interactions with specific target molecules [237]. Additionally, oxaliplatin has been shown to disrupt nucleoli through chemical modification of scaffold proteins. The disruption of this condensate led to cell death due to decreased ribosome biogenesis, which explains the mechanism of action of this commonly prescribed anti-cancer drug [238]. In another study, lipoamide and lipoic acid were found to be specifically recruited to SGs in a non-toxic manner and relieved the effects of mutant FUS in several model systems [239]. Although there is much progress to be made in the development of compounds to target phase separation, small molecules have the potential to shape the phase separation landscape.
5. Future Directions
There has been great progress in our understanding of the molecular interactions driving biomolecular condensate formation, as well as the mechanisms controlling their properties and regulation. Our understanding of the physiological roles of LLPS, as well as how corruption of LLPS is linked to disease, is also rapidly increasing. Key questions remain in our understanding of the relationship between PrLDs and LLPS. With our improved understanding of this relationship, it has become clear that the role of prions, PrLDs, and amyloidogenic proteins in LLPS must be comprehensively understood in order to understand and ultimately develop effective treatments for protein-misfolding disorders. Utilization of nucleic acids, small molecules, or chaperone proteins are promising approaches to target disordered proteins. No therapeutic treatments currently exist for prion disorders, but improved understanding of prion-like and amyloidogenic proteins has the potential to yield new advances towards the development of effective therapeutics.
Author Contributions
Conceptualization, M.L.S. and M.E.J.; Writing—Original Draft Preparation, M.L.S. and M.E.J.; Writing—Review & Editing, M.L.S. and M.E.J.; Funding Acquisition, M.L.S. and M.E.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIH grant F31NS120512 to M.L.S. and NIH grant R35GM128772, a Longer Life Foundation Award, an American Heart Association Career Development Award, and an ALS Association Award to M.E.J.
Acknowledgments
We thank members of the Jackrel Lab for feedback.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Aβ, amyloid-beta; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; α-Syn, alpha-synuclein; CPEB3, cytoplasmic polyadenylation element binding protein 3; CJD, Creutzfeldt-Jakob disease; DPRs, dipeptide repeat proteins; FTD, frontotemporal dementia; HD, Huntington’s disease; IDPs, intrinsically disordered proteins; IDRs, intrinsically disordered regions; Kap-β2, karyopherin-β2; LCDs, low complexity domains; LLPS, liquid-liquid phase separation; NDs, neurodegenerative diseases; PAR, poly(ADP-ribose); PD, Parkinson’s disease; PMEL, premelanosome protein; PrLDs, prion-like domains; PrPC, cellular prion protein; PrPSc, scrapie prion protein; PTMs, post-translation modifications; RBPs, RNA-binding proteins; SGs, stress granules; TSE, transmissible spongiform encephalopathy.
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Li, X.; Kahveci, T. A Novel algorithm for identifying low-complexity regions in a protein sequence. Bioinformatics 2006, 22, 2980–2987. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their “mysterious” (meta)physics. Front. Phys. 2019, 7. [Google Scholar] [CrossRef]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Stress, aging, and neurodegenerative disease. N. Engl. J. Med. 2006, 355, 2254–2255. [Google Scholar] [CrossRef]
- Head, M.W.; Ironside, J.W. Review: Creutzfeldt-Jakob disease: Prion protein type, disease phenotype and agent strain. Neuropathol. Appl. Neurobiol. 2012, 38, 296–310. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Jarosz, D.F.; Jones, S.K.; Chang, A.; Lancaster, A.K.; Lindquist, S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012, 482, 363–368. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 2005, 6, 435–450. [Google Scholar] [CrossRef]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Alberti, S. Prion-like low-complexity sequences: Key regulators of protein solubility and phase behavior. J. Biol. Chem. 2019, 294, 7128–7136. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Prions, chaperones, and proteostasis in yeast. Cold Spring Harb. Perspect. Biol. 2017, 9, a023663. [Google Scholar] [CrossRef]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 2018, 173, 706–719. [Google Scholar] [CrossRef]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R., 3rd; Da Cruz, S.; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371. [Google Scholar] [CrossRef]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338. [Google Scholar] [CrossRef]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Dumm, B.K.; Venus, S.; Boyko, S.; Putnam, A.A.; Jankowsky, E.; Surewicz, W.K. Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun. 2020, 11, 5574. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Brandner, S. Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2020, 46, 522–545. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Smith, J.F.; Knowles, T.P.; Dobson, C.M.; Macphee, C.E.; Welland, M.E. Characterization of the nanoscale properties of individual amyloid fibrils. Proc. Natl. Acad. Sci. USA 2006, 103, 15806–15811. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.L.; Cheng, N.; Williams, R.W.; Steven, A.C.; Wickner, R.B. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 1999, 283, 1339–1343. [Google Scholar] [CrossRef]
- Masison, D.C.; Wickner, R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef]
- Masison, D.C.; Maddelein, M.L.; Wickner, R.B. The prion model for [URE3] of yeast: Spontaneous generation and requirements for propagation. Proc. Natl. Acad. Sci. USA 1997, 94, 12503–12508. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Bezsonov, E.E.; DeWilde, M.; Ducatez, M. Yeast Prions compared to functional prions and amyloids. J. Mol. Biol. 2018, 430, 3707–3719. [Google Scholar] [CrossRef]
- Moore, R.A.; Taubner, L.M.; Priola, S.A. Prion protein misfolding and disease. Curr. Opin. Struct. Biol. 2009, 19, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.; Sidle, K.C.; Hardy, J. Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 2015, 41, 578–597. [Google Scholar] [CrossRef]
- Bistaffa, E.; Moda, F.; Virgilio, T.; Campagnani, I.; De Luca, C.M.G.; Rossi, M.; Salzano, G.; Giaccone, G.; Tagliavini, F.; Legname, G. Synthetic Prion Selection and Adaptation. Mol. Neurobiol. 2019, 56, 2978–2989. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Abid, K.; Soto, C. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta 2007, 1772, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Bessen, R.A.; McKenzie, D.; Marsh, R.F.; Aiken, J.M. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J. Virol. 2000, 74, 5542–5547. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef]
- Legname, G.; Nguyen, H.O.; Peretz, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. USA 2006, 103, 19105–19110. [Google Scholar] [CrossRef]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. Ther. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Brion, J.P.; Flament-Durand, J.; Dustin, P. Alzheimer’s disease and tau proteins. Lancet 1986, 2, 1098. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I.; Zaidi, T.; Merz, P.A.; Wen, G.Y.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective brain microtubule assembly in Alzheimer’s disease. Lancet 1986, 2, 421–426. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M.Y. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Rey, N.L.; George, S.; Steiner, J.A.; Madaj, Z.; Luk, K.C.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropath. 2018, 135, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Uemura, M.T.; Lo, A.; Bassil, F.; Zhang, B.; Luk, K.C.; Lee, V.M.; Takahashi, R.; Trojanowski, J.Q. Slow progressive accumulation of oligodendroglial alpha-synuclein (α-Syn) pathology in synthetic α-syn fibril-induced mouse models of synucleinopathy. J. Neuropathol. Exp. Neurol. 2019, 78, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, A.S.; Adler, C.H.; Serrano, G.E.; Curry, J.R.; Shill, H.A.; Kopyov, O.; Beach, T.G. Co-Existence of tau and α-synuclein pathology in fetal graft tissue at autopsy: A case report. Parkinsonism Relat. Disord. 2020, 71, 36–39. [Google Scholar] [CrossRef]
- Nekooki-Machida, Y.; Kurosawa, M.; Nukina, N.; Ito, K.; Oda, T.; Tanaka, M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9679–9684. [Google Scholar] [CrossRef]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Aslund, A.; Hammarström, P.; Nilsson, K.P.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef]
- Morozova, O.A.; March, Z.M.; Robinson, A.S.; Colby, D.W. Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 2013, 52, 6960–6967. [Google Scholar] [CrossRef]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Fowler, D.M.; Hurbain, I.; Luk, K.C.; Stayrook, S.E.; Lemmon, M.A.; Raposo, G.; Shorter, J.; Kelly, J.W.; et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 2009, 284, 35543–35555. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Drisaldi, B.; Colnaghi, L.; Fioriti, L.; Rao, N.; Myers, C.; Snyder, A.M.; Metzger, D.J.; Tarasoff, J.; Konstantinov, E.; Fraser, P.E.; et al. SUMOylation Is an Inhibitory Constraint that Regulates the Prion-like Aggregation and Activity of CPEB3. Cell Rep. 2015, 11, 1694–1702. [Google Scholar] [CrossRef]
- Fioriti, L.; Myers, C.; Huang, Y.Y.; Li, X.; Stephan, J.S.; Trifilieff, P.; Colnaghi, L.; Kosmidis, S.; Drisaldi, B.; Pavlopoulos, E.; et al. The persistence of hippocampal-based memory requires protein synthesis mediated by the prion-like protein CPEB3. Neuron 2015, 86, 1433–1448. [Google Scholar] [CrossRef]
- Pavlopoulos, E.; Trifilieff, P.; Chevaleyre, V.; Fioriti, L.; Zairis, S.; Pagano, A.; Malleret, G.; Kandel, E.R. Neuralized1 activates CPEB3: A function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell 2011, 147, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.S.; Fioriti, L.; Lamba, N.; Colnaghi, L.; Karl, K.; Derkatch, I.L.; Kandel, E.R. The CPEB3 protein is a functional prion that interacts with the actin cytoskeleton. Cell Rep. 2015, 11, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Roan, N.R.; Römling, U.; Bevins, C.L.; Münch, J. Amyloid formation: Functional friend or fearful foe? J. Int. Med. 2016, 280, 139–152. [Google Scholar] [CrossRef]
- Castellano, L.M.; Shorter, J. The surprising role of amyloid fibrils in HIV infection. Biology 2012, 1, 58–80. [Google Scholar] [CrossRef]
- Roan, N.R.; Sandi-Monroy, N.; Kohgadai, N.; Usmani, S.M.; Hamil, K.G.; Neidleman, J.; Montano, M.; Ständker, L.; Röcker, A.; Cavrois, M.; et al. Semen amyloids participate in spermatozoa selection and clearance. eLife 2017, 6. [Google Scholar] [CrossRef]
- Ho, T.; Watt, B.; Spruce, L.A.; Seeholzer, S.H.; Marks, M.S. The kringle-like domain facilitates post-endoplasmic reticulum changes to premelanosome protein (PMEL) oligomerization and disulfide bond configuration and promotes amyloid formation. J. Biol. Chem. 2016, 291, 3595–3612. [Google Scholar] [CrossRef] [PubMed]
- Si, K.; Lindquist, S.; Kandel, E.R. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003, 115, 879–891. [Google Scholar] [CrossRef]
- Khan, M.R.; Li, L.; Pérez-Sánchez, C.; Saraf, A.; Florens, L.; Slaughter, B.D.; Unruh, J.R.; Si, K. Amyloidogenic oligomerization transforms drosophila Orb2 from a translation repressor to an activator. Cell 2015, 163, 1468–1483. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Taglialegna, A.; Lasa, I.; Valle, J. Amyloid Structures as Biofilm Matrix Scaffolds. J. Bacteriol. 2016, 198, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm formation mechanisms and targets for developing antibiofilm agents. Future Med. Chem. 2015, 7, 493–512. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: Current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Khurana, V. Specification of physiologic and disease states by distinct proteins and protein conformations. Cell 2017, 171, 1001–1014. [Google Scholar] [CrossRef]
- True, H.L.; Lindquist, S.L. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef]
- Newby, G.A.; Lindquist, S. Blessings in disguise: Biological benefits of prion-like mechanisms. Trends Cell Biol. 2013, 23, 251–259. [Google Scholar] [CrossRef]
- Uptain, S.M.; Lindquist, S. Prions as protein-based genetic elements. Annu. Rev. Microbiol. 2002, 56, 703–741. [Google Scholar] [CrossRef] [PubMed]
- Tuite, M.F.; Serio, T.R. The prion hypothesis: From biological anomaly to basic regulatory mechanism. Nat. Rev. Mol. Cell Biol. 2010, 11, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Masel, J.; Siegal, M.L. Robustness: Mechanisms and consequences. Trends Genet. 2009, 25, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Baudin-Baillieu, A.; Legendre, R.; Kuchly, C.; Hatin, I.; Demais, S.; Mestdagh, C.; Gautheret, D.; Namy, O. Genome-wide translational changes induced by the prion [PSI+]. Cell Rep. 2014, 8, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Galopier, A.; Martini, C.; Matsufuji, S.; Fabret, C.; Rousset, J.P. Epigenetic control of polyamines by the prion [PSI+]. Nat. Cell Biol. 2008, 10, 1069–1075. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Kryndushkin, D.; Wickner, R.B. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. USA 2011, 108, 5337–5341. [Google Scholar] [CrossRef] [PubMed]
- Nakayashiki, T.; Kurtzman, C.P.; Edskes, H.K.; Wickner, R.B. Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 10575–10580. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Lindquist, S.L. HSP104 required for induced thermotolerance. Science 1990, 248, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Taulien, J.; Borkovich, K.A.; Lindquist, S. Hsp104 is required for tolerance to many forms of stress. EMBO J. 1992, 11, 2357–2364. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004, 304, 1793–1797. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol. Cell 2006, 23, 425–438. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008, 27, 2712–2724. [Google Scholar] [CrossRef]
- Sweeny, E.A.; Shorter, J. Prion proteostasis: Hsp104 meets its supporting cast. Prion 2008, 2, 135–140. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. PLAAC: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881–20890. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Casafont, I.; Bengoechea, R.; Tapia, O.; Berciano, M.T.; Lafarga, M. TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 2009, 167, 235–241. [Google Scholar] [CrossRef]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Müller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 2010, 5, e12247. [Google Scholar] [CrossRef]
- Ash, P.E.; Zhang, Y.J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kaneko, K.; Watanabe, S.; Yamanaka, K.; Nukina, N. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 2011, 286, 18664–18672. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220. [Google Scholar] [CrossRef] [PubMed]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.; Cone, A.; Ju, S. Supersaturated proteins in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, 5065–5066. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404, 604–609. [Google Scholar] [CrossRef]
- Platani, M.; Goldberg, I.; Swedlow, J.R.; Lamond, A.I. In vivo analysis of Cajal body movement, separation, and joining in live human cells. J. Cell Biol. 2000, 151, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Gao, Y.S.; Tousson, A.; Shah, A.; Chen, T.L.; Vertel, B.M.; Sztul, E. Nuclear aggresomes form by fusion of PML-associated aggregates. Mol. Biol. Cell 2005, 16, 4905–4917. [Google Scholar] [CrossRef]
- Chen, Y.C.; Kappel, C.; Beaudouin, J.; Eils, R.; Spector, D.L. Live cell dynamics of promyelocytic leukemia nuclear bodies upon entry into and exit from mitosis. Mol. Biol. Cell 2008, 19, 3147–3162. [Google Scholar] [CrossRef] [PubMed]
- Dellaire, G.; Ching, R.W.; Dehghani, H.; Ren, Y.; Bazett-Jones, D.P. The number of PML nuclear bodies increases in early S phase by a fission mechanism. J. Cell Sci. 2006, 119, 1026–1033. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Anastassova-Kristeva, M. The nucleolar cycle in man. J. Cell Sci. 1977, 25, 103–110. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Weber, S.C.; Brangwynne, C.P. Getting RNA and protein in phase. Cell 2012, 149, 1188–1191. [Google Scholar] [CrossRef]
- Dellaire, G.; Eskiw, C.H.; Dehghani, H.; Ching, R.W.; Bazett-Jones, D.P. Mitotic accumulations of PML protein contribute to the re-establishment of PML nuclear bodies in G1. J. Cell Sci. 2006, 119, 1034–1042. [Google Scholar] [CrossRef]
- Grousl, T.; Ivanov, P.; Frýdlová, I.; Vasicová, P.; Janda, F.; Vojtová, J.; Malínská, K.; Malcová, I.; Nováková, L.; Janosková, D.; et al. Robust heat shock induces eIF2alpha-phosphorylation-independent assembly of stress granules containing eIF3 and 40S ribosomal subunits in budding yeast, Saccharomyces cerevisiae. J. Cell Sci. 2009, 122, 2078–2088. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, N.P.; Castelli, L.M.; Campbell, S.G.; Holmes, L.E.; Ashe, M.P. Stress-dependent relocalization of translationally primed mRNPs to cytoplasmic granules that are kinetically and spatially distinct from P-bodies. J. Cell Biol. 2007, 179, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Louria-Hayon, I.; Grossman, T.; Sionov, R.V.; Alsheich, O.; Pandolfi, P.P.; Haupt, Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J. Biol. Chem. 2003, 278, 33134–33141. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Mathieu, C.; Kolaitis, R.M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a tunable switch that triggers phase separation to assemble stress granules. Cell 2020, 181, 325–345. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlüßler, R.; Kim, K.; Trussina, I.; Wang, J.; Mateju, D.; et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 2020, 181, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J., Jr.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H., Jr.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [PubMed]
- Crick, S.L.; Ruff, K.M.; Garai, K.; Frieden, C.; Pappu, R.V. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 20075–20080. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Mannen, T.; Yamashita, S.; Tomita, K.; Goshima, N.; Hirose, T. The Sam68 nuclear body is composed of two RNase-sensitive substructures joined by the adaptor HNRNPL. J. Cell Biol. 2016, 214, 45–59. [Google Scholar] [CrossRef]
- Boke, E.; Mitchison, T.J. The balbiani body and the concept of physiological amyloids. Cell Cycle 2017, 16, 153–154. [Google Scholar] [CrossRef][Green Version]
- Boke, E.; Ruer, M.; Wühr, M.; Coughlin, M.; Lemaitre, R.; Gygi, S.P.; Alberti, S.; Drechsel, D.; Hyman, A.A.; Mitchison, T.J. Amyloid-like self-assembly of a cellular compartment. Cell 2016, 166, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.O.; Passos, Y.M.; do Amaral, M.J.; Macedo, B.; Tempone, M.H.; Bezerra, O.C.L.; Moraes, M.O.; Almeida, M.S.; Weber, G.; Missailidis, S.; et al. Liquid-liquid phase separation and fibrillation of the prion protein modulated by a high-affinity DNA aptamer. FASEB J. 2020, 34, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Zhang, H.; Elbaum-Garfinkle, S.; Langdon, E.M.; Taylor, N.; Occhipinti, P.; Bridges, A.A.; Brangwynne, C.P.; Gladfelter, A.S. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell 2015, 60, 220–230. [Google Scholar] [CrossRef]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef]
- Martinez, F.J.; Pratt, G.A.; Van Nostrand, E.L.; Batra, R.; Huelga, S.C.; Kapeli, K.; Freese, P.; Chun, S.J.; Ling, K.; Gelboin-Burkhart, C.; et al. Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron 2016, 92, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 2018, 173, 693–705. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., 3rd; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Lee, Y.-B.; Chen, H.-J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 2016, 167, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Mugler, C.F.; Hondele, M.; Heinrich, S.; Sachdev, R.; Vallotton, P.; Koek, A.Y.; Chan, L.Y.; Weis, K. ATPase activity of the DEAD-box protein Dhh1 controls processing body formation. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef] [PubMed]
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235. [Google Scholar] [CrossRef]
- Frottin, F.; Schueder, F.; Tiwary, S.; Gupta, R.; Körner, R.; Schlichthaerle, T.; Cox, J.; Jungmann, R.; Hartl, F.U.; Hipp, M.S. The nucleolus functions as a phase-separated protein quality control compartment. Science 2019, 365, 342–347. [Google Scholar] [CrossRef]
- DeSantis, M.E.; Leung, E.H.; Sweeny, E.A.; Jackrel, M.E.; Cushman-Nick, M.; Neuhaus-Follini, A.; Vashist, S.; Sochor, M.A.; Knight, M.N.; Shorter, J. Operational plasticity enables Hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012, 151, 778–793. [Google Scholar] [CrossRef]
- Gates, S.N.; Yokom, A.L.; Lin, J.; Jackrel, M.E.; Rizo, A.N.; Kendsersky, N.M.; Buell, C.E.; Sweeny, E.A.; Mack, K.L.; Chuang, E.; et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 357, 273–279. [Google Scholar] [CrossRef]
- Yokom, A.L.; Gates, S.N.; Jackrel, M.E.; Mack, K.L.; Su, M.; Shorter, J.; Southworth, D.R. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat. Struct. Mol. Biol. 2016, 23, 830–837. [Google Scholar] [CrossRef]
- Parsell, D.A.; Kowal, A.S.; Singer, M.A.; Lindquist, S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994, 372, 475–478. [Google Scholar] [CrossRef]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82. [Google Scholar] [CrossRef]
- Glover, J.R.; Tkach, J.M. Crowbars and ratchets: Hsp100 chaperones as tools in reversing protein aggregation. Biochem. Cell Biol. 2001, 79, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Jackrel, M.E.; DeSantis, M.E.; Martinez, B.A.; Castellano, L.M.; Stewart, R.M.; Caldwell, K.A.; Caldwell, G.A.; Shorter, J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014, 156, 170–182. [Google Scholar] [CrossRef]
- Jackrel, M.E.; Shorter, J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis. Models Mech. 2014, 7, 1175–1184. [Google Scholar] [CrossRef]
- Jackrel, M.E.; Shorter, J. Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 2015, 9, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Jackrel, M.E.; Yee, K.; Tariq, A.; Chen, A.I.; Shorter, J. Disparate mutations confer therapeutic gain of Hsp104 function. ACS Chem. Biol. 2015, 10, 2672–2679. [Google Scholar] [CrossRef]
- Ryan, J.J.; Sprunger, M.L.; Holthaus, K.; Shorter, J.; Jackrel, M.E. Engineered protein disaggregases mitigate toxicity of aberrant prion-like fusion proteins underlying sarcoma. J. Biol. Chem. 2019, 294, 11286–11296. [Google Scholar] [CrossRef]
- Jackrel, M.E.; Shorter, J. Reversing deleterious protein aggregation with re-engineered protein disaggregases. Cell Cycle 2014, 13, 1379–1383. [Google Scholar] [CrossRef]
- Jackrel, M.E.; Shorter, J. Protein-Remodeling factors as potential therapeutics for neurodegenerative disease. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Tariq, A.; Lin, J.; Jackrel, M.E.; Hesketh, C.D.; Carman, P.J.; Mack, K.L.; Weitzman, R.; Gambogi, C.; Hernandez Murillo, O.A.; Sweeny, E.A.; et al. Mining disaggregase sequence space to safely counter TDP-43, FUS, and α-synuclein proteotoxicity. Cell Rep. 2019, 28, 2080–2095. [Google Scholar] [CrossRef]
- Yasuda, K.; Clatterbuck-Soper, S.F.; Jackrel, M.E.; Shorter, J.; Mili, S. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol. 2017, 216, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Bao, A.; Bell, B.; Ling, C.; Jackrel, M.E. Drivers of Hsp104 potentiation revealed by scanning mutagenesis of the middle domain. Protein Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Torrente, M.P.; Chuang, E.; Noll, M.M.; Jackrel, M.E.; Go, M.S.; Shorter, J. Mechanistic insights into Hsp104 potentiation. J. Biol. Chem. 2016, 291, 5101–5115. [Google Scholar] [CrossRef]
- Tariq, A.; Lin, J.; Noll, M.M.; Torrente, M.P.; Mack, K.L.; Murillo, O.H.; Jackrel, M.E.; Shorter, J. Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS Yeast Res. 2018, 18. [Google Scholar] [CrossRef]
- Michalska, K.; Zhang, K.; March, Z.M.; Hatzos-Skintges, C.; Pintilie, G.; Bigelow, L.; Castellano, L.M.; Miles, L.J.; Jackrel, M.E.; Chuang, E.; et al. Structure of Calcarisporiella thermophila Hsp104 disaggregase that antagonizes diverse proteotoxic misfolding events. Structure 2019, 27, 449–463. [Google Scholar] [CrossRef]
- March, Z.M.; Sweeney, K.; Kim, H.; Yan, X.; Castellano, L.M.; Jackrel, M.E.; Lin, J.; Chuang, E.; Gomes, E.; Willicott, C.W.; et al. Therapeutic genetic variation revealed in diverse Hsp104 homologs. eLife 2020, 9. [Google Scholar] [CrossRef]
- Vacher, C.; Garcia-Oroz, L.; Rubinsztein, D.C. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2005, 14, 3425–3433. [Google Scholar] [CrossRef]
- Lo Bianco, C.; Shorter, J.; Régulier, E.; Lashuel, H.; Iwatsubo, T.; Lindquist, S.; Aebischer, P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Investig. 2008, 118, 3087–3097. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef] [PubMed]
- Poepsel, S.; Sprengel, A.; Sacca, B.; Kaschani, F.; Kaiser, M.; Gatsogiannis, C.; Raunser, S.; Clausen, T.; Ehrmann, M. Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 2015, 11, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G.; et al. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLoS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef] [PubMed]
- Torrente, M.P.; Shorter, J. The metazoan protein disaggregase and amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat shock proteins. Prion 2013, 7, 457–463. [Google Scholar] [CrossRef]
- Zhu, G.; Harischandra, D.S.; Ghaisas, S.; Zhang, P.; Prall, W.; Huang, L.; Maghames, C.; Guo, L.; Luna, E.; Mack, K.L.; et al. TRIM11 Prevents and Reverses Protein Aggregation and Rescues a Mouse Model of Parkinson’s Disease. Cell Rep. 2020, 33, 108418. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R. P-bodies and stress granules: Possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol. 2012, 4, a012286. [Google Scholar] [CrossRef]
- Saha, S.; Weber, C.A.; Nousch, M.; Adame-Arana, O.; Hoege, C.; Hein, M.Y.; Osborne-Nishimura, E.; Mahamid, J.; Jahnel, M.; Jawerth, L.; et al. Polar positioning of phase-separated liquid compartments in cells regulated by an mRNA competition mechanism. Cell 2016, 166, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.; Weber, S.C.; Vaidya, N.; Haataja, M.; Brangwynne, C.P. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E5237–E5245. [Google Scholar] [CrossRef] [PubMed]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef]
- Wei, M.T.; Elbaum-Garfinkle, S.; Holehouse, A.S.; Chen, C.C.; Feric, M.; Arnold, C.B.; Priestley, R.D.; Pappu, R.V.; Brangwynne, C.P. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 2017, 9, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.T.; Bonomi, M.; Vendruscolo, M. Structural Ensemble modulation upon small-molecule binding to disordered proteins. J. Mol. Biol. 2018, 430, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Drug startups coalesce around condensates. Nat. Biotechnol. 2021, 39, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Updike, D.L.; Hachey, S.J.; Kreher, J.; Strome, S. P granules extend the nuclear pore complex environment in the C. elegans germ line. J. Cell Biol. 2011, 192, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Hammoudeh, D.I.; Daab, A.T.; Metallo, S.J. Small-molecule perturbation of competing interactions between c-Myc and Max. Bioorg. Med. Chem. Lett. 2009, 19, 807–810. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.T.; Aprile, F.A.; Michaels, T.C.T.; Limbocker, R.; Perni, M.; Ruggeri, F.S.; Mannini, B.; Löhr, T.; Bonomi, M.; Camilloni, C.; et al. Small-molecule sequestration of amyloid-β as a drug discovery strategy for Alzheimer’s disease. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Wyman, J.; Gill, S.J. Ligand-linked phase changes in a biological system: Applications to sickle cell hemoglobin. Proc. Natl. Acad. Sci. USA 1980, 77, 5239–5242. [Google Scholar] [CrossRef]
- Posey, A.E.; Ruff, K.M.; Harmon, T.S.; Crick, S.L.; Li, A.; Diamond, M.I.; Pappu, R.V. Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. J. Biol. Chem. 2018, 293, 3734–3746. [Google Scholar] [CrossRef]
- Ruff, K.M.; Dar, F.; Pappu, R.V. Ligand effects on phase separation of multivalent macromolecules. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Park, H.J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef]
- Wheeler, R.J. Therapeutics-how to treat phase separation-associated diseases. Emerg. Top. Life Sci. 2020, 4, 307–318. [Google Scholar] [CrossRef]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grofte, M.; Rask, M.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef]
- Klein, I.A.; Boija, A.; Afeyan, L.K.; Hawken, S.W.; Fan, M.; Dall’Agnese, A.; Oksuz, O.; Henninger, J.E.; Shrinivas, K.; Sabari, B.R.; et al. Partitioning of cancer therapeutics in nuclear condensates. Science 2020, 368, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.B.; Jafaar, Z.; Rodencal, J.J.; Leonetti, M.D.; Dixon, S.J.; Rohatgi, R.; Brandman, O. Oxaliplatin kills cells via liquid-liquid demixing of nucleoli. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wheeler, R.J.; Lee, H.O.; Poser, I.; Pal, A.; Doeleman, T.; Kishigami, S.; Kour, S.; Anderson, E.N.; Marrone, L.; Murthy, A.C.; et al. Small molecules for modulating protein driven liquid-liquid phase separation in treating neurodegenerative disease. bioRxiv 2019. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).