
Intrinsic Disorder and Phosphorylation in BRCA2 Facilitate Tight Regulation of Multiple Conserved Binding Events

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
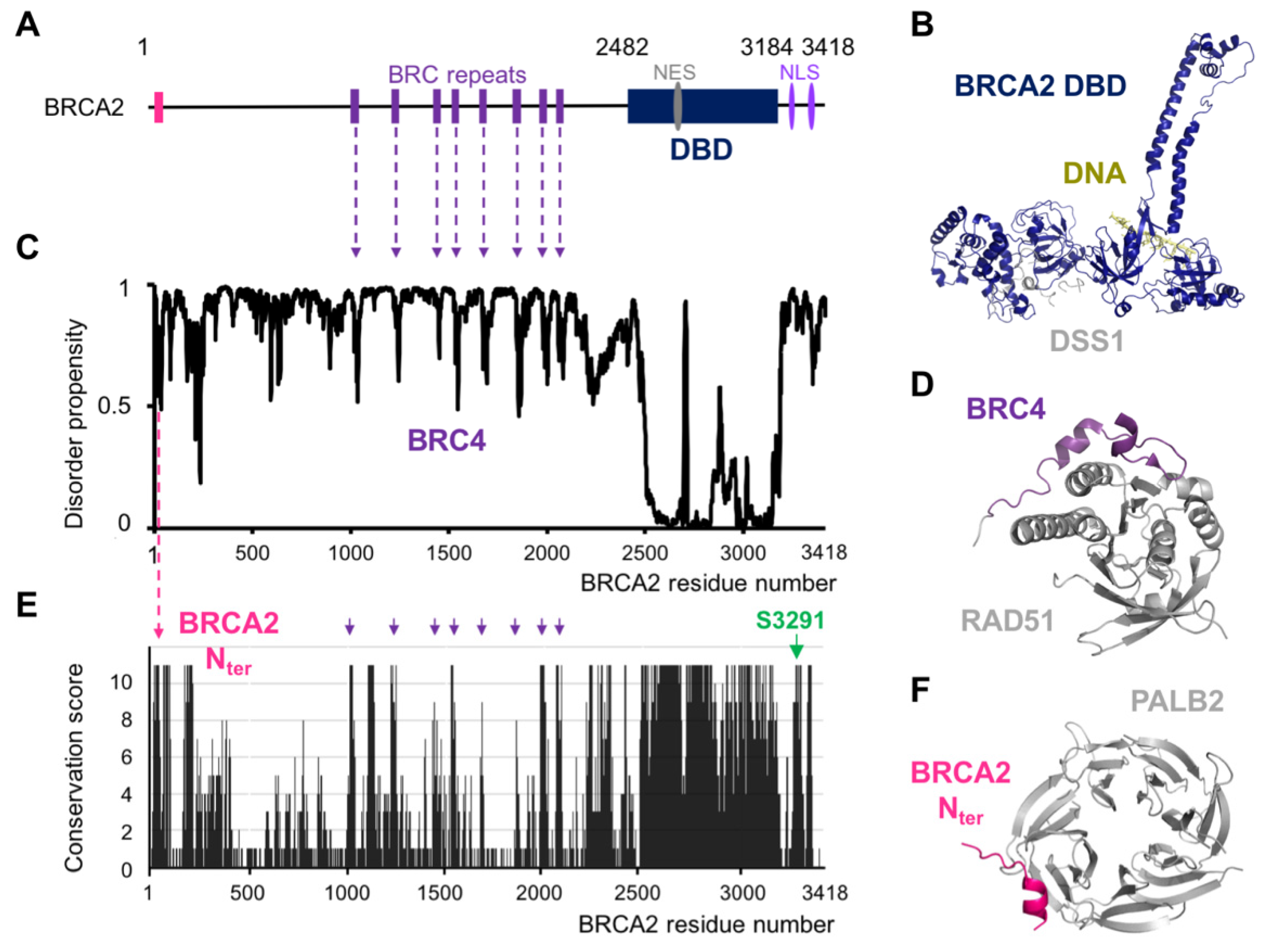
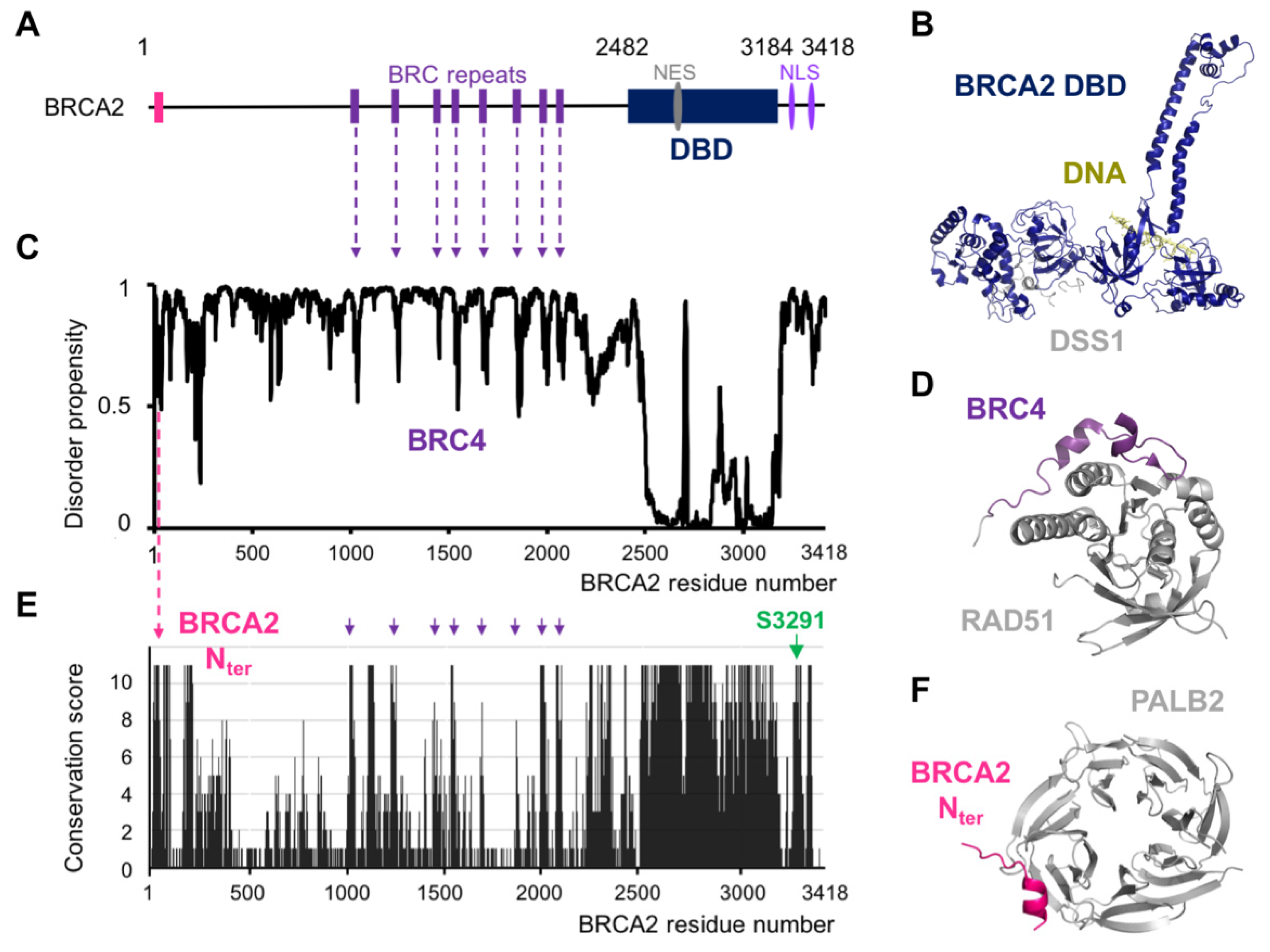
2. BRCA2 Exhibits a Set of Conserved Motifs Predicted to Be Disordered
3. Structural Studies Illustrate the Role of BRCA2 Disordered Regions in DNA Repair by HR
4. During Interphase, the C-Terminal Disordered Region of BRCA2 Is Required for the Protection of Stalled Forks
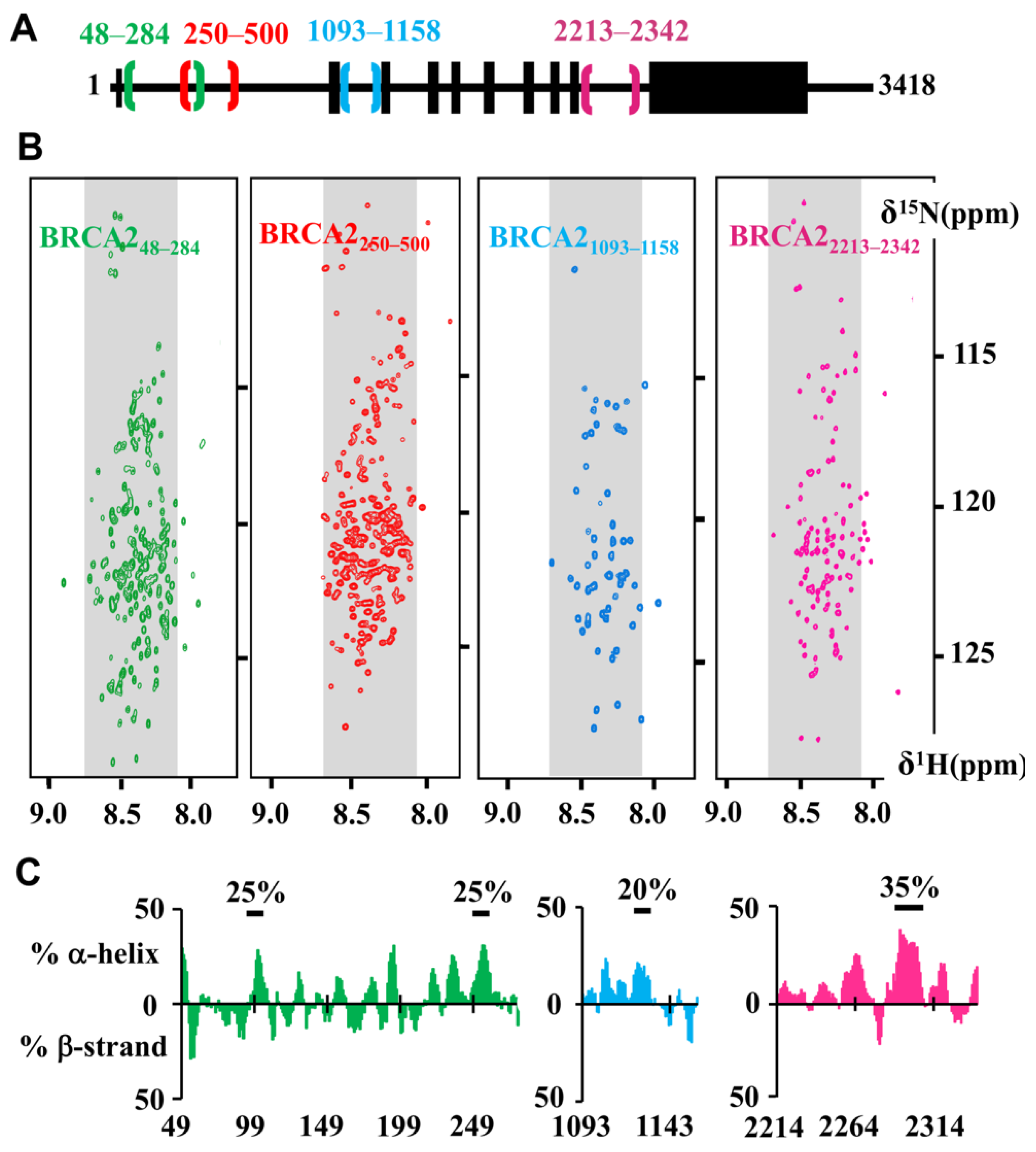
5. Nuclear Magnetic Resonance Analyses Support the Presence of Additional Disordered and Conserved Regions in BRCA2
6. BRCA248–284 Phosphorylation by CDKs and PLK1 Ensures Correct DNA Repair before Mitosis and BRCA2 Midbody Localization during Cytokinesis
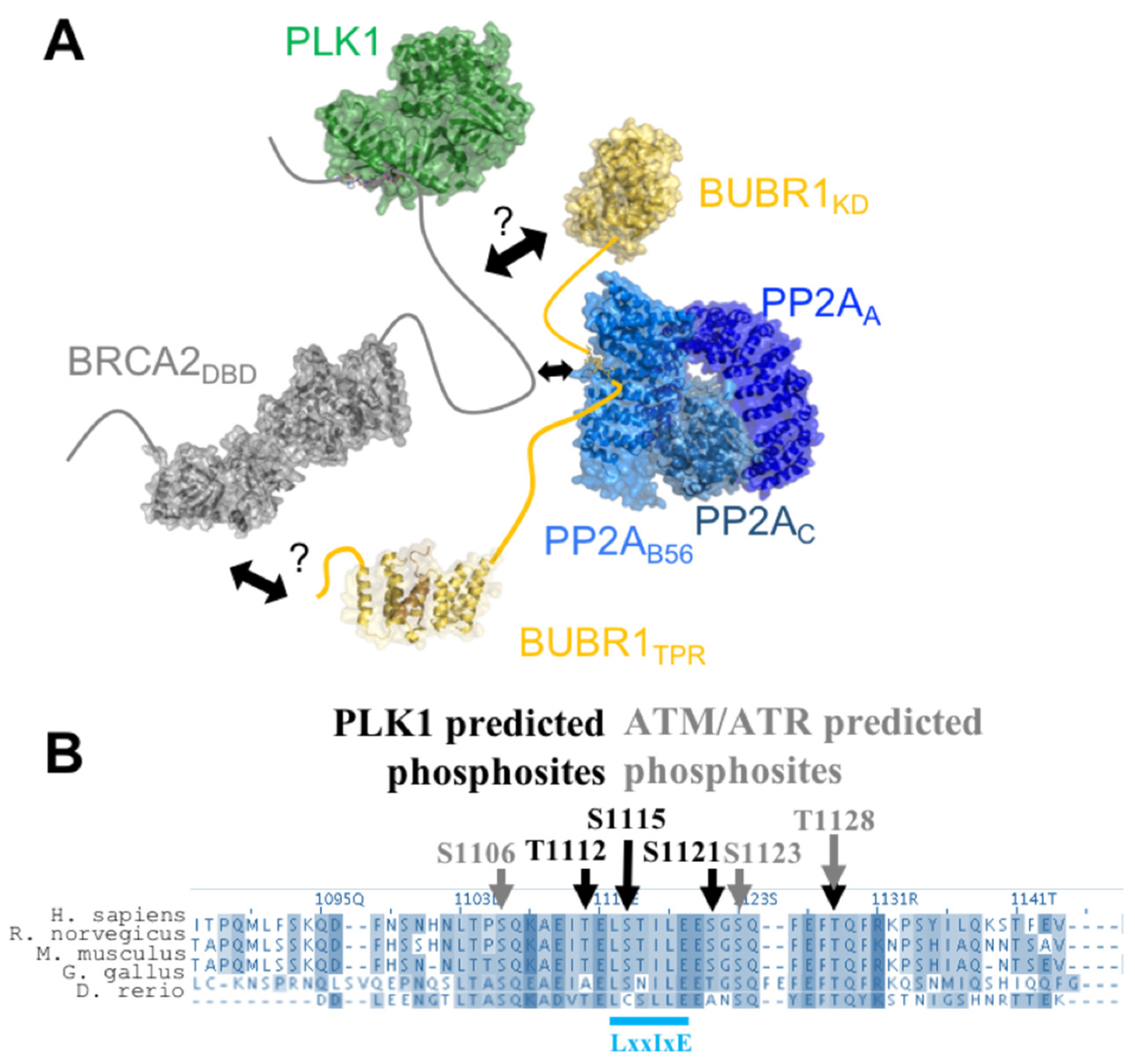
7. BRCA2199–210 and BRCA21093–1158 Mediate the Assembly of a Large Complex, Including PLK1, BUBR1, and PP2A, Regulating Chromosome Alignment during Mitosis
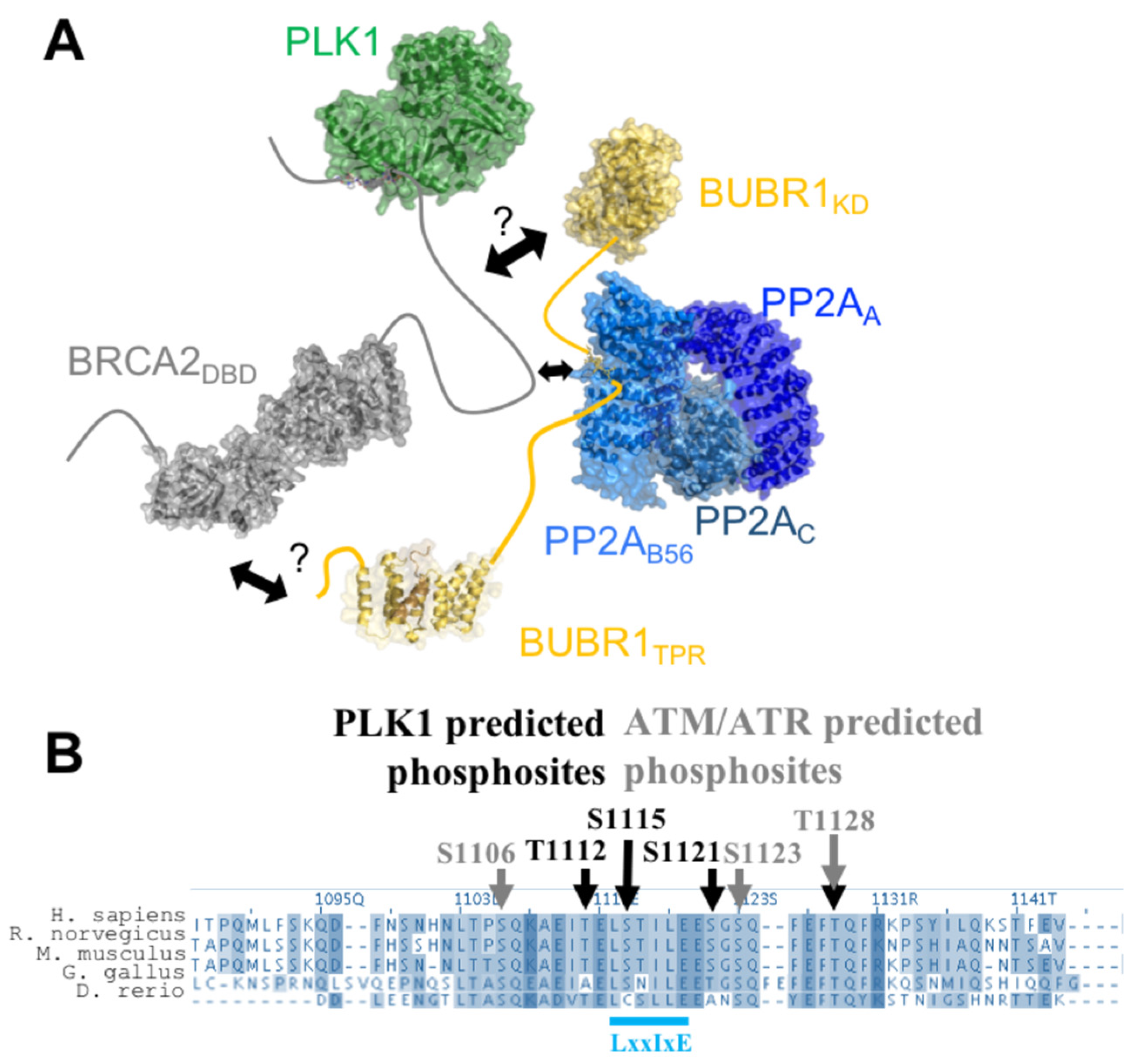
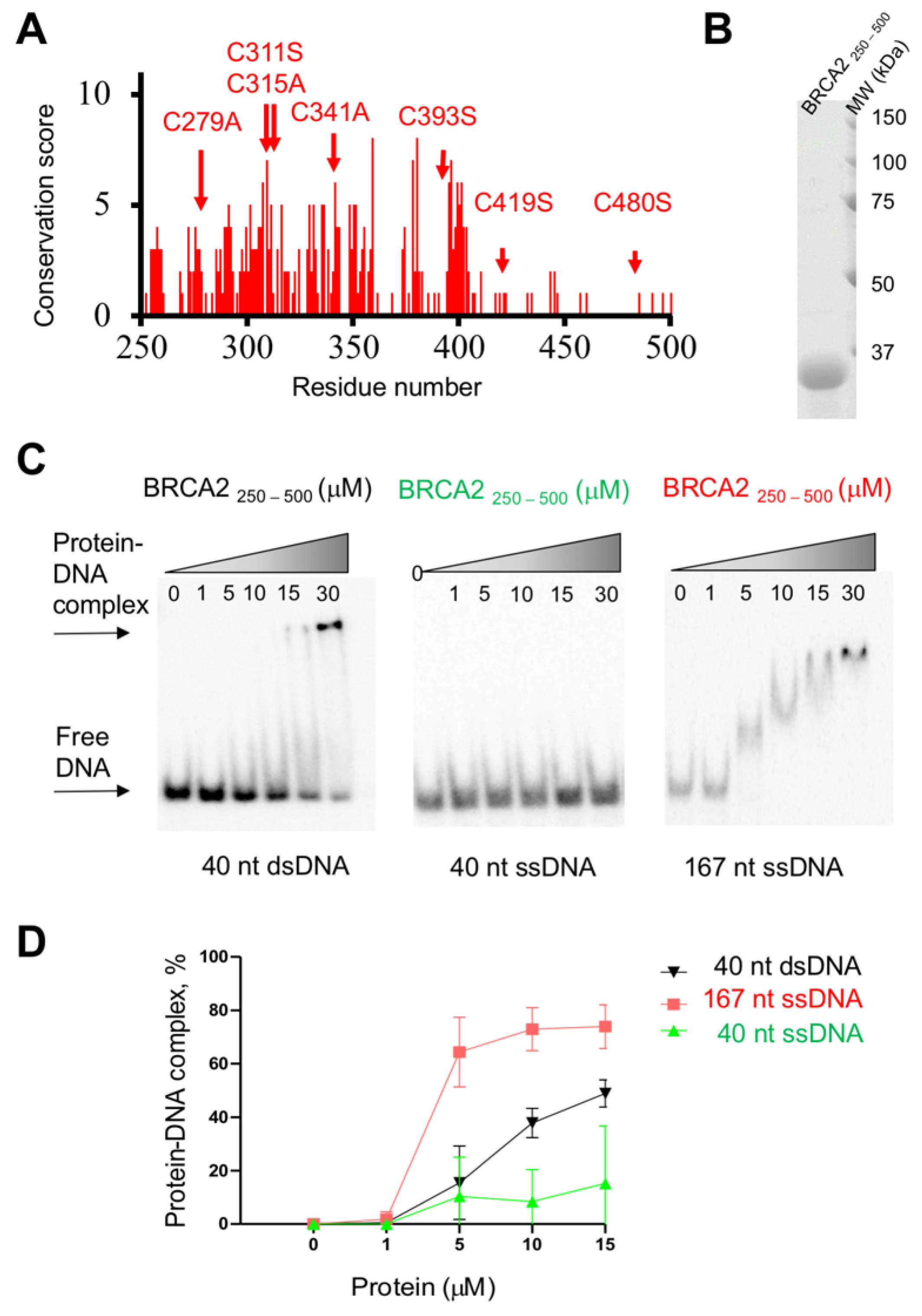
8. Both Folded and Disordered Regions of BRCA2 Contribute to Its DNA Binding Properties
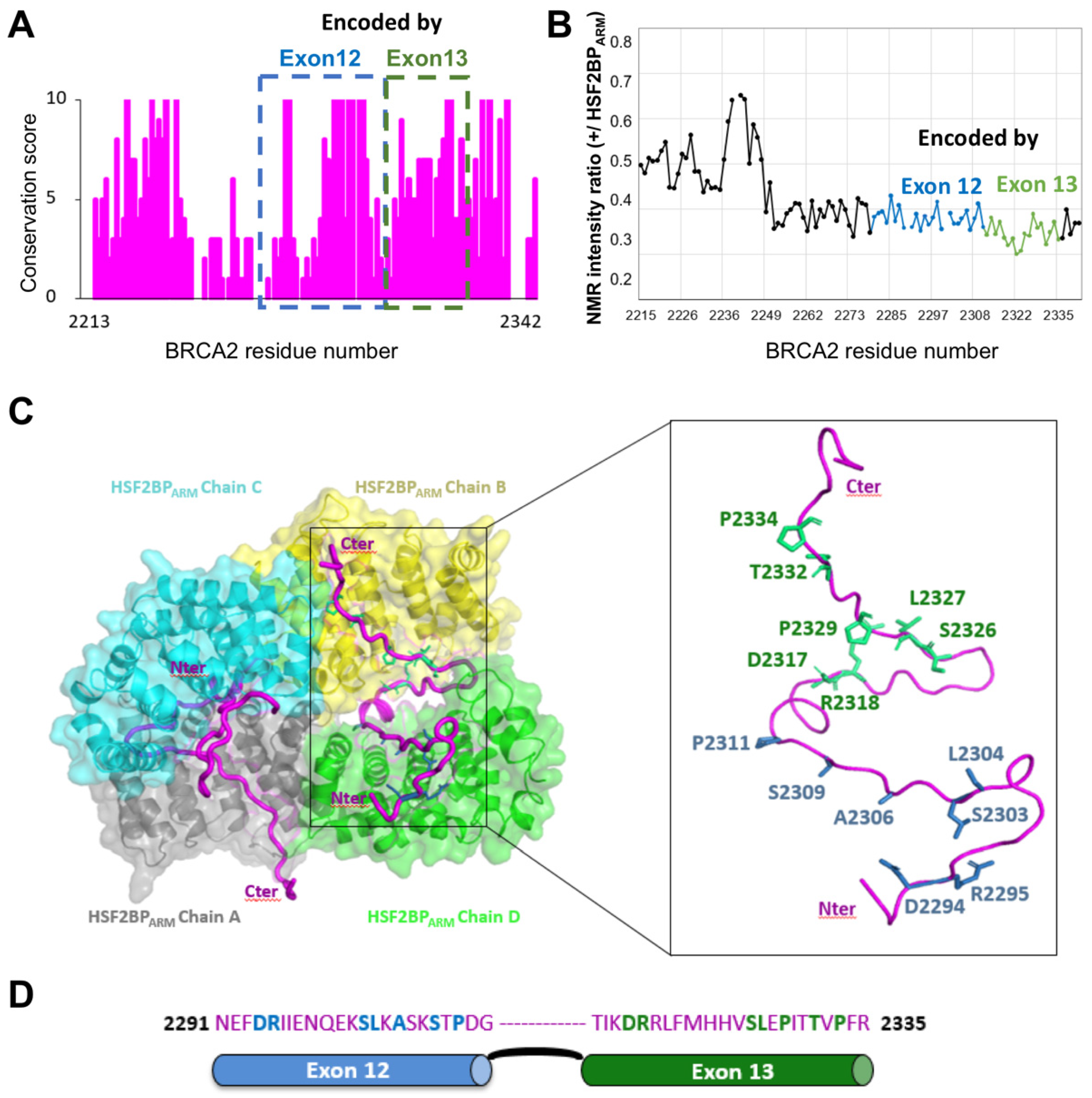
9. A Conserved and Disordered BRCA2 Region Contains a Cryptic Repeat That Recruits HSF2BP, Thus Triggering BRCA2 Degradation
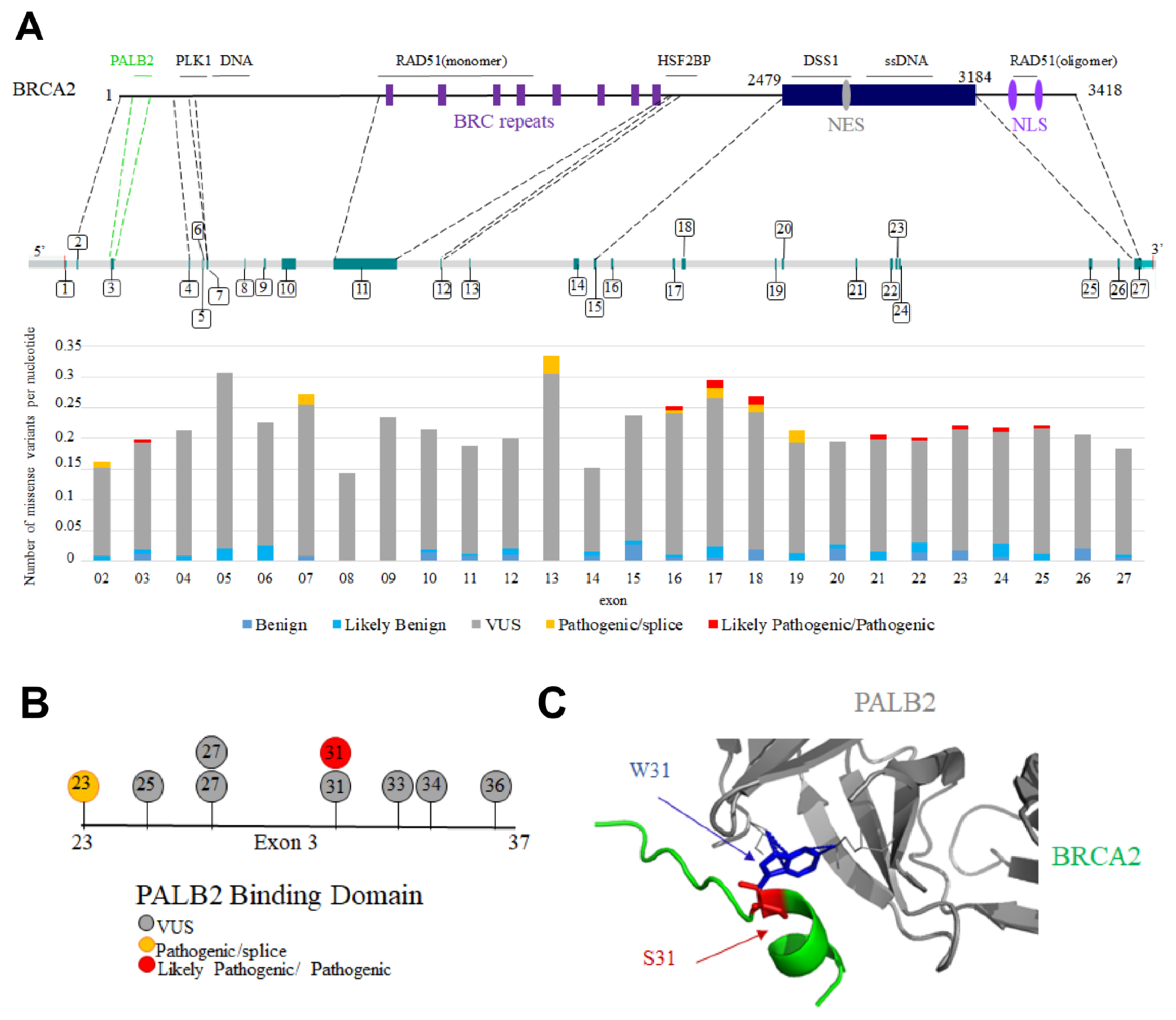
10. BRCA2 Variants Detected in Patients with Cancers Are Mutated in Folded or Disordered Regions: Which of These Variants Are Pathogenic?

11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Baker, M.D. Analysis of DNA repair and recombination responses in mouse cells depleted for Brca2 by SiRNA. DNA Repair 2007, 6, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U.; et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Choi, E.S.; Hwang, K.; Kim, J.; Sampath, S.; Venkitaraman, A.R.; Lee, H. The breast cancer susceptibility gene BRCA2 is required for the maintenance of telomere homeostasis. J. Biol. Chem. 2012, 287, 5091–5101. [Google Scholar] [CrossRef] [Green Version]
- Daniels, M.J.; Wang, Y.; Lee, M.; Venkitaraman, A.R. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science 2004, 306, 876–879. [Google Scholar] [CrossRef]
- Takaoka, M.; Saito, H.; Takenaka, K.; Miki, Y.; Nakanishi, A. BRCA2 phosphorylated by PLK1 moves to the midbody to regulate cytokinesis mediated by nonmuscle myosin IIC. Cancer Res. 2014, 74, 1518–1528. [Google Scholar] [CrossRef] [Green Version]
- Sharan, S.K.; Pyle, A.; Coppola, V.; Babus, J.; Swaminathan, S.; Benedict, J.; Swing, D.; Martin, B.K.; Tessarollo, L.; Evans, J.P.; et al. BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development 2004, 131, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.K.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Trainer, A.H.; Friedman, L.S.; Thistlethwaite, F.C.; Evans, M.J.; Ponder, B.A.; Venkitaraman, A.R. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol. Cell 1999, 4, 1–10. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef]
- Szabo, C.I.; King, M.C. Inherited breast and ovarian cancer. Hum. Mol. Genet. 1995, 4, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thoma, N.H.; Zheng, N.; Chen, P.L.; Lee, W.H.; Pavletich, N.P. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.; Paliwal, K.K.; Litfin, T.; Zhou, Y. SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning. Genom. Proteom. Bioinform. 2019, 17, 645–656. [Google Scholar] [CrossRef]
- Shahid, T.; Soroka, J.; Kong, E.; Malivert, L.; McIlwraith, M.J.; Pape, T.; West, S.C.; Zhang, X. Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat. Struct. Mol. Biol. 2014, 21, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, H.; Paul, M.W.; Grosbart, M.; van Rossum-Fikkert, S.E.; Lebbink, J.H.G.; Kanaar, R.; Houtsmuller, A.B.; Wyman, C. Architectural plasticity of human BRCA2-RAD51 complexes in DNA break repair. Nucleic Acids Res. 2017, 45, 4507–4518. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Grosbart, M.; Sanchez, H.; Verhagen, B.; van der Zon, N.L.L.; Ristic, D.; van Rossum-Fikkert, S.E.; Wyman, C. Conformational flexibility and oligomerization of BRCA2 regions induced by RAD51 interaction. Nucleic Acids Res. 2020, 48, 9649–9659. [Google Scholar] [CrossRef] [PubMed]
- Le, H.P.; Ma, X.; Vaquero, J.; Brinkmeyer, M.; Guo, F.; Heyer, W.D.; Liu, J. DSS1 and ssDNA regulate oligomerization of BRCA2. Nucleic Acids Res. 2020, 48, 7818–7833. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Tompa, P.; Simon, I.; Dosztanyi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Functional advantages of dynamic protein disorder. FEBS Lett. 2015, 589, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [Green Version]
- Moesa, H.A.; Wakabayashi, S.; Nakai, K.; Patil, A. Chemical composition is maintained in poorly conserved intrinsically disordered regions and suggests a means for their classification. Mol. Biosyst. 2012, 8, 3262–3273. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.T.; Mulder, F.A.A. Quality and bias of protein disorder predictors. Sci. Rep. 2019, 9, 5137. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.S.; von Nicolai, C.; Kim, T.; Ehlen, A.; Mazin, A.V.; Kowalczykowski, S.C.; Carreira, A. BRCA2 regulates DMC1-mediated recombination through the BRC repeats. Proc. Natl. Acad. Sci. USA 2016, 113, 3515–3520. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 1993, 9, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.J.; Yu, V.P.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.; Venkitaraman, A.R. Involvement of Brca2 in DNA repair. Mol. Cell 1998, 1, 347–357. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Park, J.Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Vaithiyalingam, S.; San Filippo, J.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef] [Green Version]
- Dray, E.; Siaud, N.; Dubois, E.; Doutriaux, M.P. Interaction between Arabidopsis Brca2 and its partners Rad51, Dmc1, and Dss1. Plant Physiol. 2006, 140, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorslund, T.; Esashi, F.; West, S.C. Interactions between human BRCA2 protein and the meiosis-specific recombinase DMC1. EMBO J. 2007, 26, 2915–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef]
- Davies, O.R.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat. Struct Mol. Biol. 2007, 14, 475–483. [Google Scholar] [CrossRef]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005, 434, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Niraj, J.; Pauty, J.; Maity, R.; Zhao, W.; Coulombe, Y.; Sung, P.; Masson, J.Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014, 6, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, N.; Rajendra, E.; Su, X.; Jeyasekharan, A.D.; Mahen, R.; Venkitaraman, A.R. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr. Biol. 2009, 19, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Julien, M.; Miron, S.; Carreira, A.; Theillet, F.X.; Zinn-Justin, S. (1)H, (13)C and (15)N backbone resonance assignment of the human BRCA2 N-terminal region. Biomol. NMR Assign. 2020, 14, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Ghouil, R.; Miron, S.; Koornneef, L.; Veerman, J.; Paul, M.W.; Le Du, M.H.; Sleddens, E.; van Rossum-Fikkert, S.E.; van Loon, Y.; Felipe-Medina, N.; et al. BRCA2 binding through a cryptic repeated motif to HSF2BP oligomers does not impact meiotic recombination. BioRxiv 2020. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. NMR illuminates intrinsic disorder. Curr. Opin. Struct. Biol. 2021, 70, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tamiola, K.; Mulder, F.A. Using NMR chemical shifts to calculate the propensity for structural order and disorder in proteins. Biochem. Soc. Trans. 2012, 40, 1014–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlen, A.; Martin, C.; Miron, S.; Julien, M.; Theillet, F.X.; Ropars, V.; Sessa, G.; Beaurepere, R.; Boucherit, V.; Duchambon, P.; et al. Proper chromosome alignment depends on BRCA2 phosphorylation by PLK1. Nat. Commun. 2020, 11, 1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.R.; Ting, N.S.; Qin, J.; Lee, W.H. M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex. J. Biol. Chem. 2003, 278, 35979–35987. [Google Scholar] [CrossRef] [Green Version]
- Yata, K.; Bleuyard, J.Y.; Nakato, R.; Ralf, C.; Katou, Y.; Schwab, R.A.; Niedzwiedz, W.; Shirahige, K.; Esashi, F. BRCA2 coordinates the activities of cell-cycle kinases to promote genome stability. Cell Rep. 2014, 7, 1547–1559. [Google Scholar] [CrossRef] [Green Version]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef]
- Mondal, G.; Rowley, M.; Guidugli, L.; Wu, J.; Pankratz, V.S.; Couch, F.J. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell 2012, 23, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, A.E.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Suijkerbuijk, S.J.; Vleugel, M.; Teixeira, A.; Kops, G.J. Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev. Cell 2012, 23, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Bajaj, R.; Bollen, M.; Peti, W.; Page, R. Expanding the PP2A Interactome by Defining a B56-Specific SLiM. Structure 2016, 24, 2174–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.G.; Chen, H.; Guo, F.; Yadav, V.K.; McIlwain, S.J.; Rowse, M.; Choudhary, A.; Lin, Z.; Li, Y.; Gu, T.; et al. PP2A-B’ holoenzyme substrate recognition, regulation and role in cytokinesis. Cell Discov. 2017, 3, 17027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, E.P.T.; Kruse, T.; Davey, N.E.; Lopez-Mendez, B.; Sigurethsson, J.O.; Montoya, G.; Olsen, J.V.; Nilsson, J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol. Cell. 2016, 63, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Foley, E.A.; Maldonado, M.; Kapoor, T.M. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat. Cell Biol. 2011, 13, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Futamura, M.; Arakawa, H.; Matsuda, K.; Katagiri, T.; Saji, S.; Miki, Y.; Nakamura, Y. Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBR1. Cancer Res. 2000, 60, 1531–1535. [Google Scholar]
- Choi, E.; Park, P.G.; Lee, H.O.; Lee, Y.K.; Kang, G.H.; Lee, J.W.; Han, W.; Lee, H.C.; Noh, D.Y.; Lekomtsev, S.; et al. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Dev. Cell 2012, 22, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Z.; Yu, T.; Yang, H.; Virshup, D.M.; Kops, G.J.; Lee, S.H.; Zhou, W.; Li, X.; Xu, W.; et al. Crystal structure of a PP2A B56-BubR1 complex and its implications for PP2A substrate recruitment and localization. Protein Cell 2016, 7, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Ambjoern, S.M.; Duxin, J.P.; Hertz, E.P.T.; Nasa, I.; Duro, J.; Kruse, T.; Mendez, B.L.; Rymarczyk, B.; Cressey, L.E.; van Overeem Hansen, T.; et al. A complex of BRCA2 and PP2A-B56 is required for DNA repair by homologous recombination. BioRxiv 2021. [Google Scholar] [CrossRef]
- Lee, M.; Daniels, M.J.; Venkitaraman, A.R. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 2004, 23, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Von Nicolai, C.; Ehlen, A.; Martin, C.; Zhang, X.; Carreira, A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, I.; Sato, K.; van Rossum-Fikkert, S.E.; van Vliet, N.; Sleddens, E.; Reuter, M.; Odijk, H.; van den Tempel, N.; Dekkers, D.H.W.; Bezstarosti, K.; et al. HSF2BP Interacts with a Conserved Domain of BRCA2 and Is Required for Mouse Spermatogenesis. Cell Rep. 2019, 27, 3790–3798.e7. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Brandsma, I.; van Rossum-Fikkert, S.E.; Verkaik, N.; Oostra, A.B.; Dorsman, J.C.; van Gent, D.C.; Knipscheer, P.; Kanaar, R.; Zelensky, A.N. HSF2BP negatively regulates homologous recombination in DNA interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 2442–2456. [Google Scholar] [CrossRef]
- Zhang, J.; Fujiwara, Y.; Yamamoto, S.; Shibuya, H. A meiosis-specific BRCA2 binding protein recruits recombinases to DNA double-strand breaks to ensure homologous recombination. Nat. Commun. 2019, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gurusaran, M.; Fujiwara, Y.; Zhang, K.; Echbarthi, M.; Vorontsov, E.; Guo, R.; Pendlebury, D.F.; Alam, I.; Livera, G.; et al. The BRCA2-MEILB2-BRME1 complex governs meiotic recombination and impairs the mitotic BRCA2-RAD51 function in cancer cells. Nat. Commun. 2020, 11, 2055. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Caputo, S.; Benboudjema, L.; Sinilnikova, O.; Rouleau, E.; Beroud, C.; Lidereau, R.; French, B.G.G.C.C. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012, 40, D992–D1002. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, F.; Mebirouk, N.; Jiao, Y.; Barjhoux, L.; Belotti, M.; Laurent, M.; Leone, M.; Houdayer, C.; Bressac-de Paillerets, B.; Vaur, D.; et al. GEMO, a National Resource to Study Genetic Modifiers of Breast and Ovarian Cancer Risk in BRCA1 and BRCA2 Pathogenic Variant Carriers. Front. Oncol. 2018, 8, 490. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [Green Version]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadalo, L.; Aalfs, C.M.; Agata, S.; Aittomaki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef] [Green Version]
- Mesman, R.L.S.; Calleja, F.; de la Hoya, M.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. Alternative mRNA splicing can attenuate the pathogenicity of presumed loss-of-function variants in BRCA2. Genet. Med. 2020, 22, 1355–1365. [Google Scholar] [CrossRef]
- Rosenthal, E.; Moyes, K.; Arnell, C.; Evans, B.; Wenstrup, R.J. Incidence of BRCA1 and BRCA2 non-founder mutations in patients of Ashkenazi Jewish ancestry. Breast Cancer Res. Treat. 2015, 149, 223–227. [Google Scholar] [CrossRef]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef]
- Muller, D.; Rouleau, E.; Schultz, I.; Caputo, S.; Lefol, C.; Bieche, I.; Caron, O.; Nogues, C.; Limacher, J.M.; Demange, L.; et al. An entire exon 3 germ-line rearrangement in the BRCA2 gene: Pathogenic relevance of exon 3 deletion in breast cancer predisposition. BMC Med. Genet. 2011, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Caputo, S.M.; Leone, M.; Damiola, F.; Ehlen, A.; Carreira, A.; Gaidrat, P.; Martins, A.; Brandao, R.D.; Peixoto, A.; Vega, A.; et al. Full in-frame exon 3 skipping of BRCA2 confers high risk of breast and/or ovarian cancer. Oncotarget 2018, 9, 17334–17348. [Google Scholar] [CrossRef]
- Tubeuf, H.; Caputo, S.M.; Sullivan, T.; Rondeaux, J.; Krieger, S.; Caux-Moncoutier, V.; Hauchard, J.; Castelain, G.; Fievet, A.; Meulemans, L.; et al. Calibration of Pathogenicity Due to Variant-Induced Leaky Splicing Defects by Using BRCA2 Exon 3 as a Model System. Cancer Res. 2020, 80, 3593–3605. [Google Scholar] [CrossRef]
- Gaildrat, P.; Krieger, S.; Di Giacomo, D.; Abdat, J.; Revillion, F.; Caputo, S.; Vaur, D.; Jamard, E.; Bohers, E.; Ledemeney, D.; et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J. Med. Genet. 2012, 49, 609–617. [Google Scholar] [CrossRef]
- Thirthagiri, E.; Klarmann, K.D.; Shukla, A.K.; Southon, E.; Biswas, K.; Martin, B.K.; North, S.L.; Magidson, V.; Burkett, S.; Haines, D.C.; et al. BRCA2 minor transcript lacking exons 4-7 supports viability in mice and may account for survival of humans with a pathogenic biallelic mutation. Hum. Mol. Genet. 2016, 25, 1934–1945. [Google Scholar] [CrossRef] [Green Version]
- Stauffer, S.; Biswas, K.; Sharan, S.K. Bypass of premature stop codons and generation of functional BRCA2 by exon skipping. J. Hum. Genet. 2020, 65, 805–809. [Google Scholar] [CrossRef]
- Di Giacomo, D.; Gaildrat, P.; Abuli, A.; Abdat, J.; Frebourg, T.; Tosi, M.; Martins, A. Functional analysis of a large set of BRCA2 exon 7 variants highlights the predictive value of hexamer scores in detecting alterations of exonic splicing regulatory elements. Hum. Mutat. 2013, 34, 1547–1557. [Google Scholar] [CrossRef]
- Li, L.; Biswas, K.; Habib, L.A.; Kuznetsov, S.G.; Hamel, N.; Kirchhoff, T.; Wong, N.; Armel, S.; Chong, G.; Narod, S.A.; et al. Functional redundancy of exon 12 of BRCA2 revealed by a comprehensive analysis of the c.6853A>G (p.I2285V) variant. Hum. Mutat. 2009, 30, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meulemans, L.; Mesman, R.L.S.; Caputo, S.M.; Krieger, S.; Guillaud-Bataille, M.; Caux-Moncoutier, V.; Leone, M.; Boutry-Kryza, N.; Sokolowska, J.; Revillion, F.; et al. Skipping Nonsense to Maintain Function: The Paradigm of BRCA2 Exon 12. Cancer Res. 2020, 80, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.; Neuhausen, S.L.; Hansen, T.V.; Couch, F.J.; Vreeswijk, M.P.; et al. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toland, A.E.; Andreassen, P.R. DNA repair-related functional assays for the classification of BRCA1 and BRCA2 variants: A critical review and needs assessment. J. Med. Genet. 2017, 54, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Kohsaka, S.; Ueno, T.; Momozawa, Y.; Inoue, S.; Tamura, K.; Shimomura, A.; Hosoya, N.; Kobayashi, H.; Tanaka, S.; et al. High-throughput functional evaluation of BRCA2 variants of unknown significance. Nat. Commun. 2020, 11, 2573. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.E.; Hu, C.; Lee, K.Y.; LaDuca, H.; Fulk, K.; Durda, K.M.; Deckman, A.M.; Goldgar, D.E.; Monteiro, A.N.A.; Gnanaolivu, R.; et al. Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance. Am. J. Hum. Genet. 2021, 108, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Joergensen, A.M.; Howlett, N.G.; Lisby, M.; Oestergaard, V.H. A distinct role for recombination repair factors in an early cellular response to transcription-replication conflicts. Nucleic Acids Res. 2020, 48, 5467–5484. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Sessa, G.; Gomez-Gonzalez, B.; Silva, S.; Perez-Calero, C.; Beaurepere, R.; Barroso, S.; Martineau, S.; Martin, C.; Ehlen, A.; Martinez, J.S.; et al. BRCA2 promotes DNA-RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repairdouble dagger. EMBO J. 2021, 40, e106018. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, H.; Mesman, R.L.S.; Von Nicolai, C.; Ehlen, A.; Guidugli, L.; Martin, C.; Calleja, F.; Meeks, H.; Hallberg, E.; Hinton, J.; et al. BRCA2 Hypomorphic Missense Variants Confer Moderate Risks of Breast Cancer. Cancer Res. 2017, 77, 2789–2799. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Julien, M.; Ghouil, R.; Petitalot, A.; Caputo, S.M.; Carreira, A.; Zinn-Justin, S. Intrinsic Disorder and Phosphorylation in BRCA2 Facilitate Tight Regulation of Multiple Conserved Binding Events. Biomolecules 2021, 11, 1060. https://doi.org/10.3390/biom11071060
Julien M, Ghouil R, Petitalot A, Caputo SM, Carreira A, Zinn-Justin S. Intrinsic Disorder and Phosphorylation in BRCA2 Facilitate Tight Regulation of Multiple Conserved Binding Events. Biomolecules. 2021; 11(7):1060. https://doi.org/10.3390/biom11071060
Chicago/Turabian StyleJulien, Manon, Rania Ghouil, Ambre Petitalot, Sandrine M. Caputo, Aura Carreira, and Sophie Zinn-Justin. 2021. "Intrinsic Disorder and Phosphorylation in BRCA2 Facilitate Tight Regulation of Multiple Conserved Binding Events" Biomolecules 11, no. 7: 1060. https://doi.org/10.3390/biom11071060
APA StyleJulien, M., Ghouil, R., Petitalot, A., Caputo, S. M., Carreira, A., & Zinn-Justin, S. (2021). Intrinsic Disorder and Phosphorylation in BRCA2 Facilitate Tight Regulation of Multiple Conserved Binding Events. Biomolecules, 11(7), 1060. https://doi.org/10.3390/biom11071060