
Multiomic Approach to Analyze Infant Gut Microbiota: Experimental and Analytical Method Optimization

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
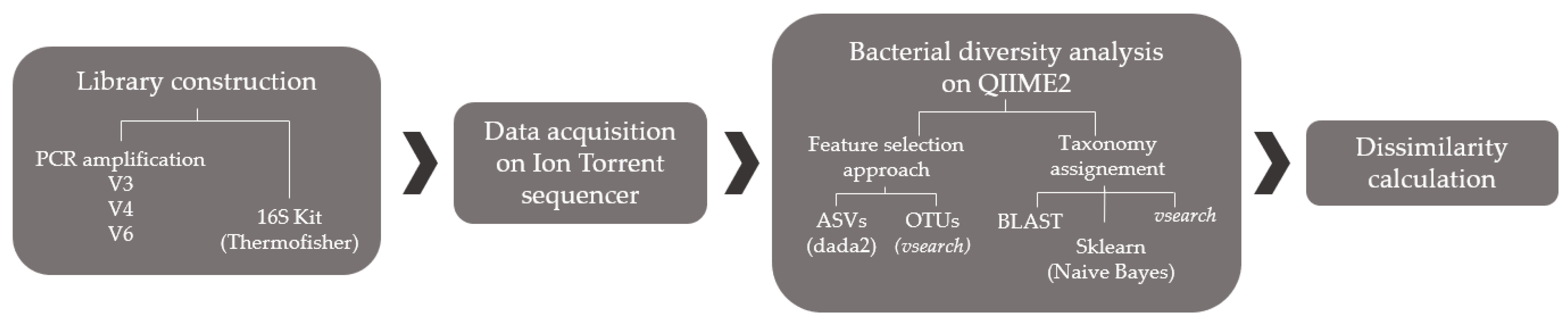
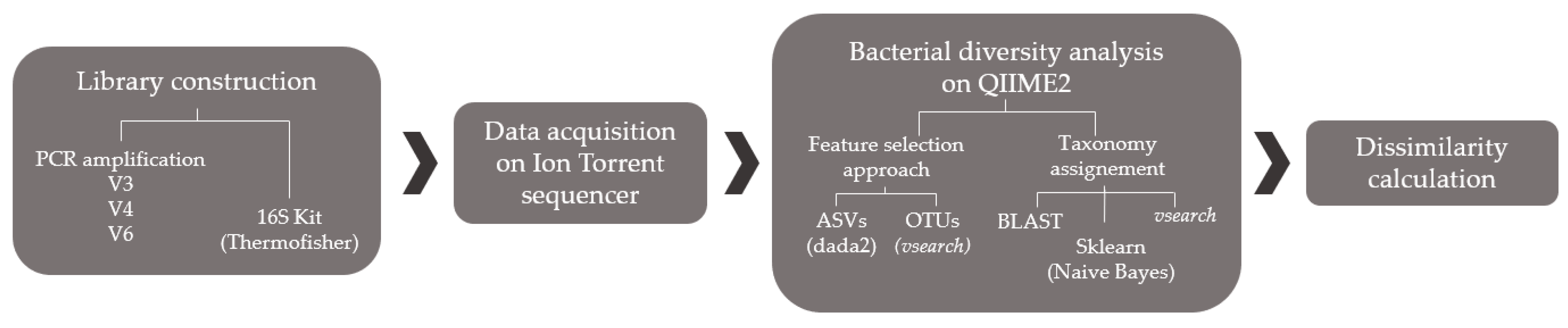
2.1. Method Optimization
2.1.1. Bacterial Strains and DNA Preparation
2.1.2. Library Preparation
Tag-Fusion Primers
Ion 16s Metagenomic Kit from Life Technologies
2.1.3. Ion Torrent PGM Sequencing
2.1.4. QIIME2 Analysis
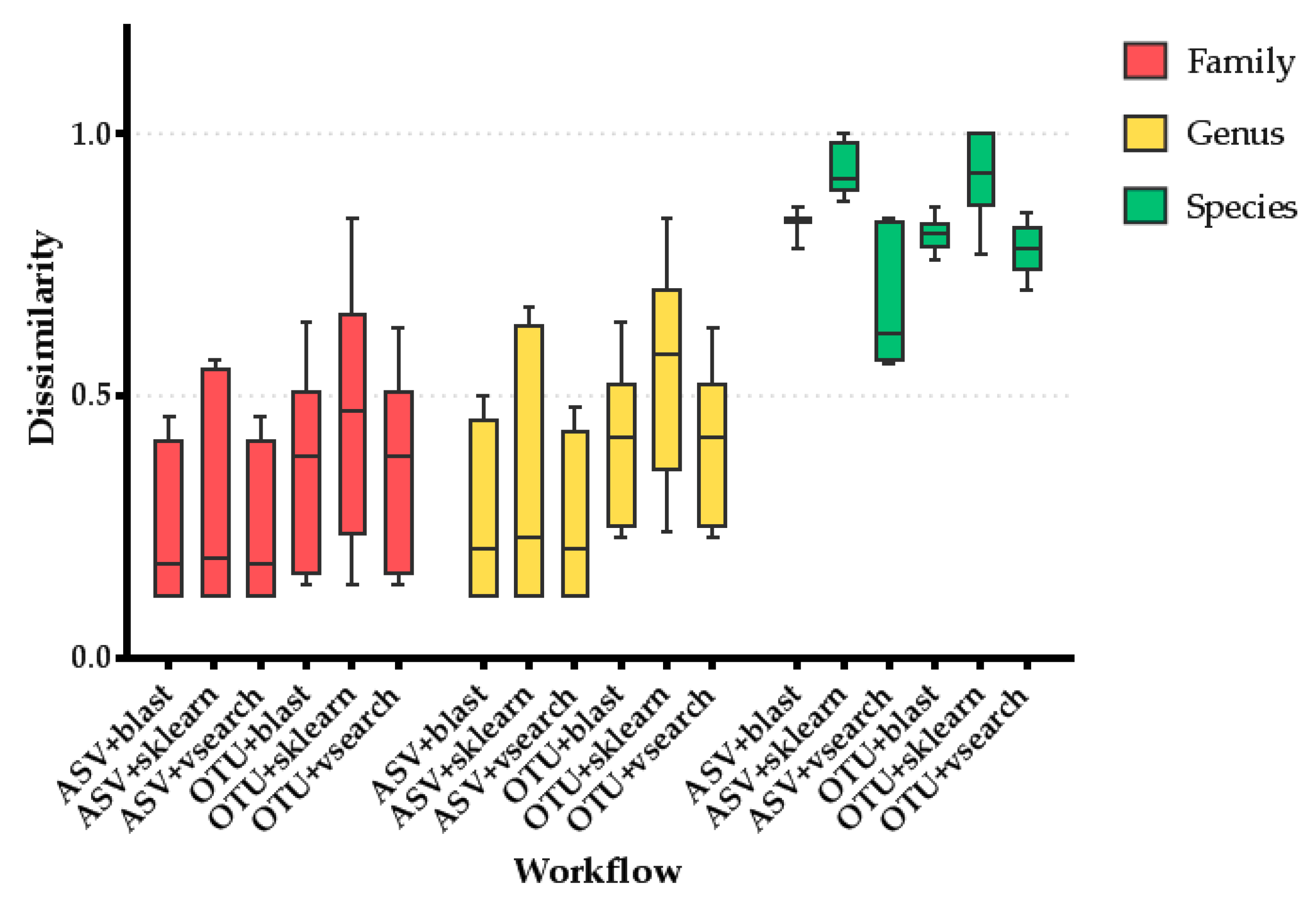
2.1.5. Performance Comparison
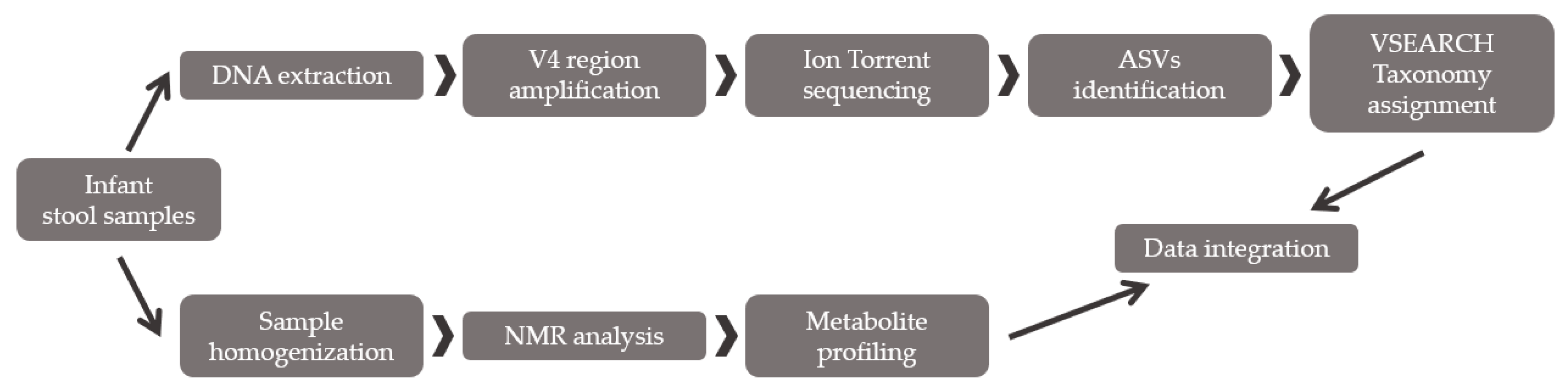
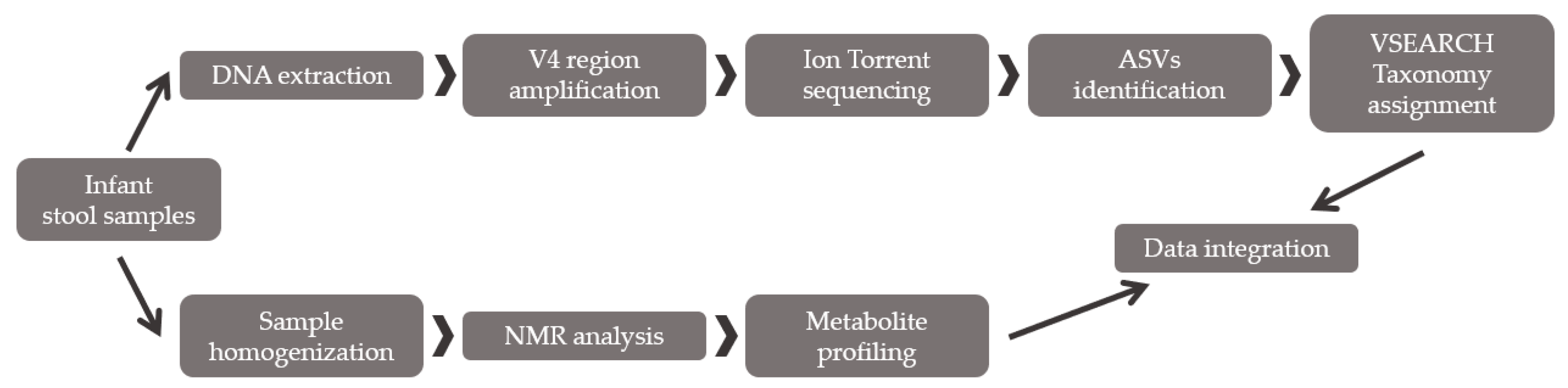
2.2. Method Validation
2.2.1. Fecal Bacterial Diversity Analysis through Partial 16S rRNA Sequencing
2.2.2. Fecal Metabolite Profiling by NMR
2.2.3. Integration between Metabolites and Gut Bacterial Composition
3. Results
3.1. Method Optimization
3.2. Method Validation
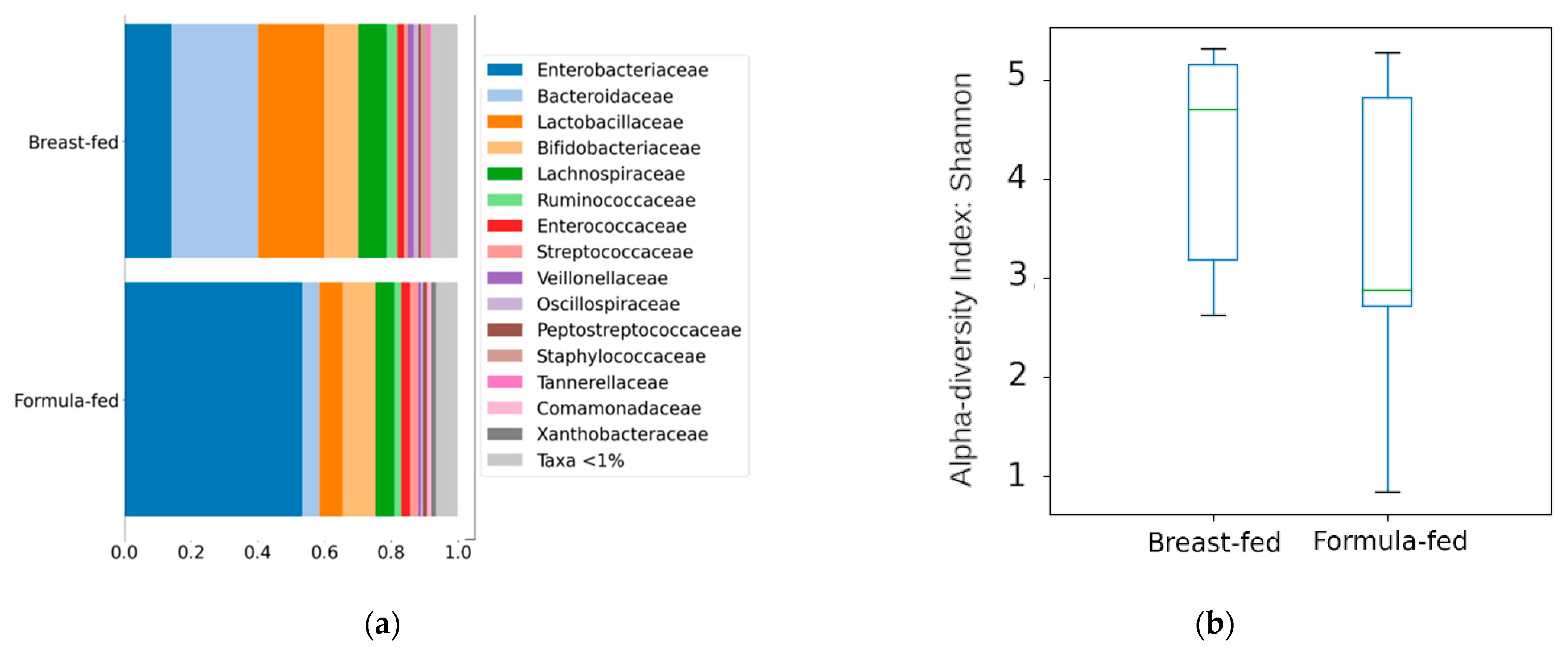
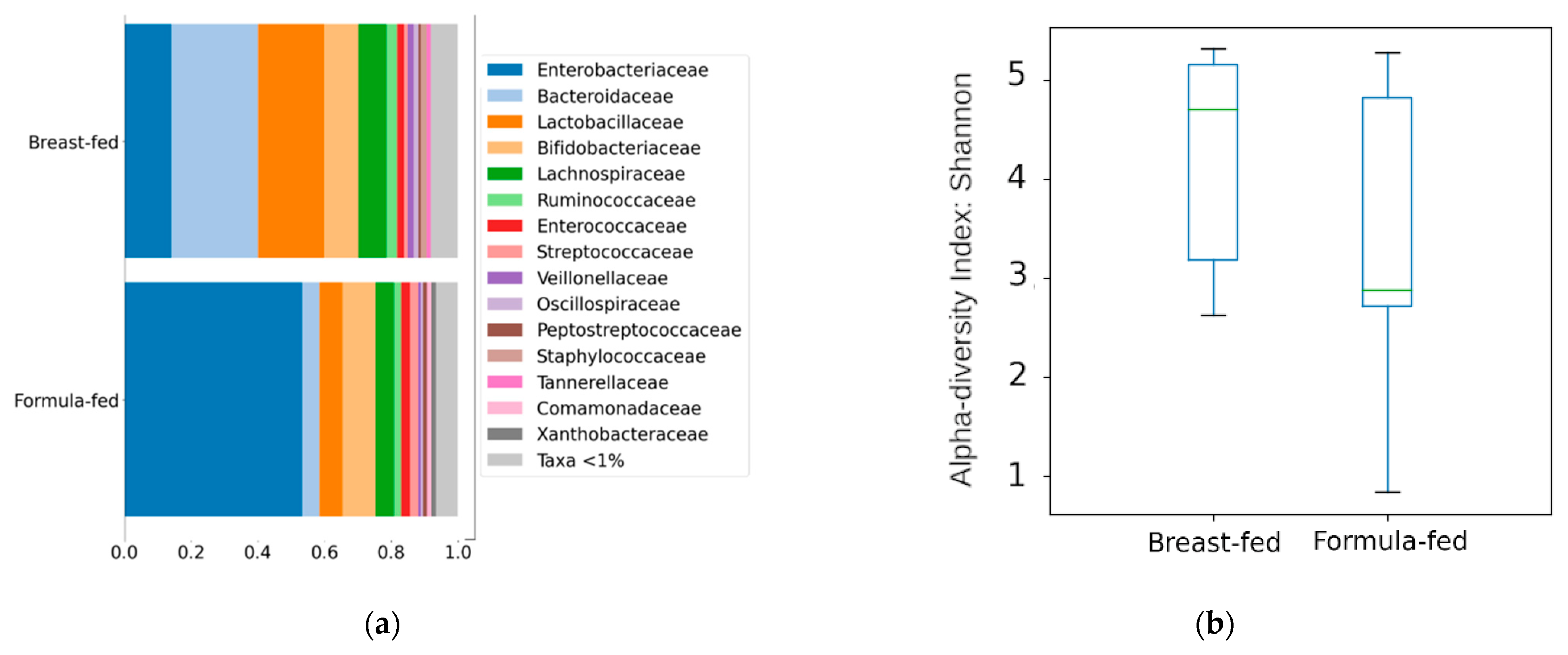
3.2.1. Differences in Bacterial Diversity between the Experimental Groups
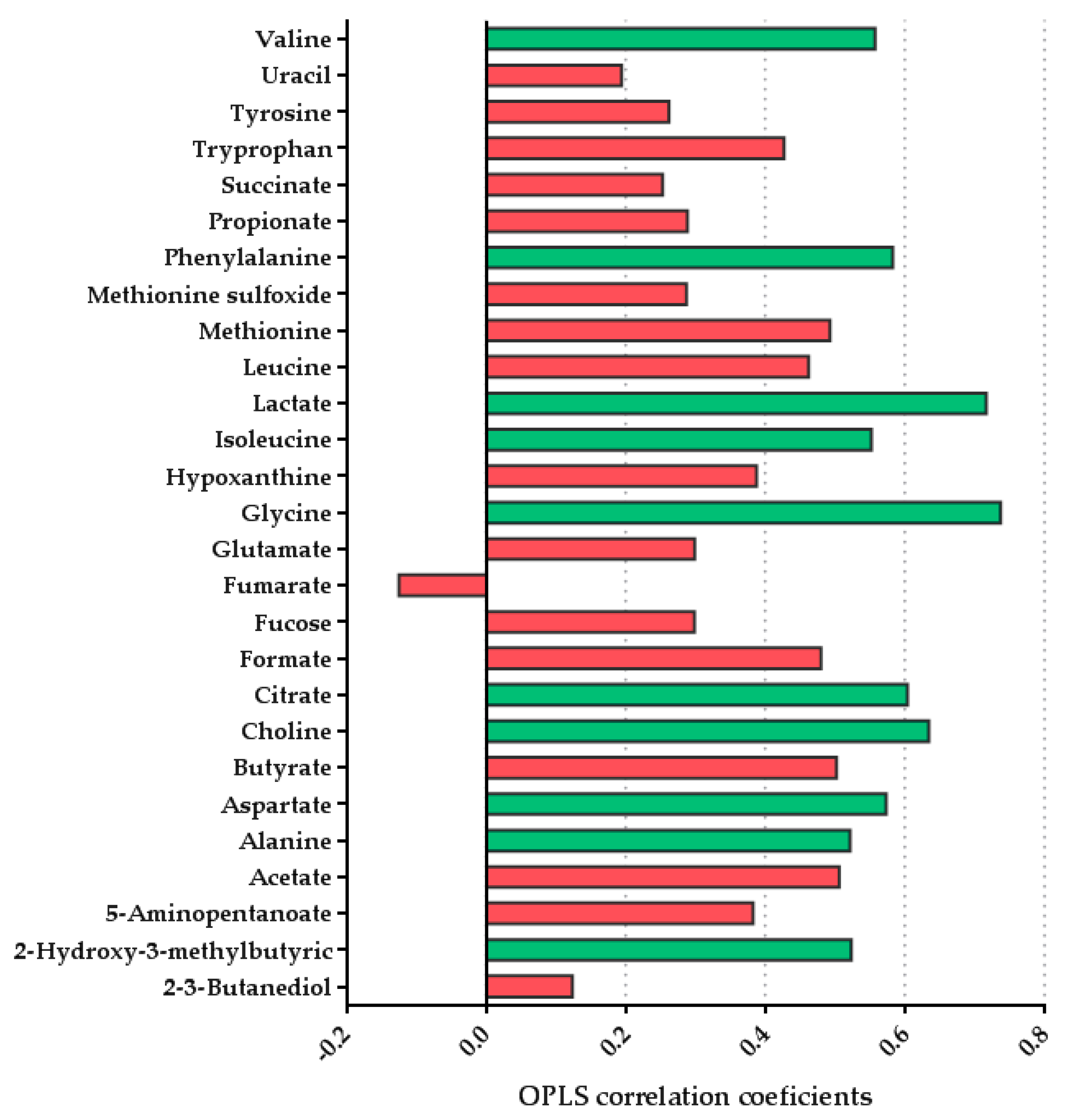
3.2.2. Multivariate Analysis Showed Differences in the Fecal Metabolome Depending on the Type of Feeding
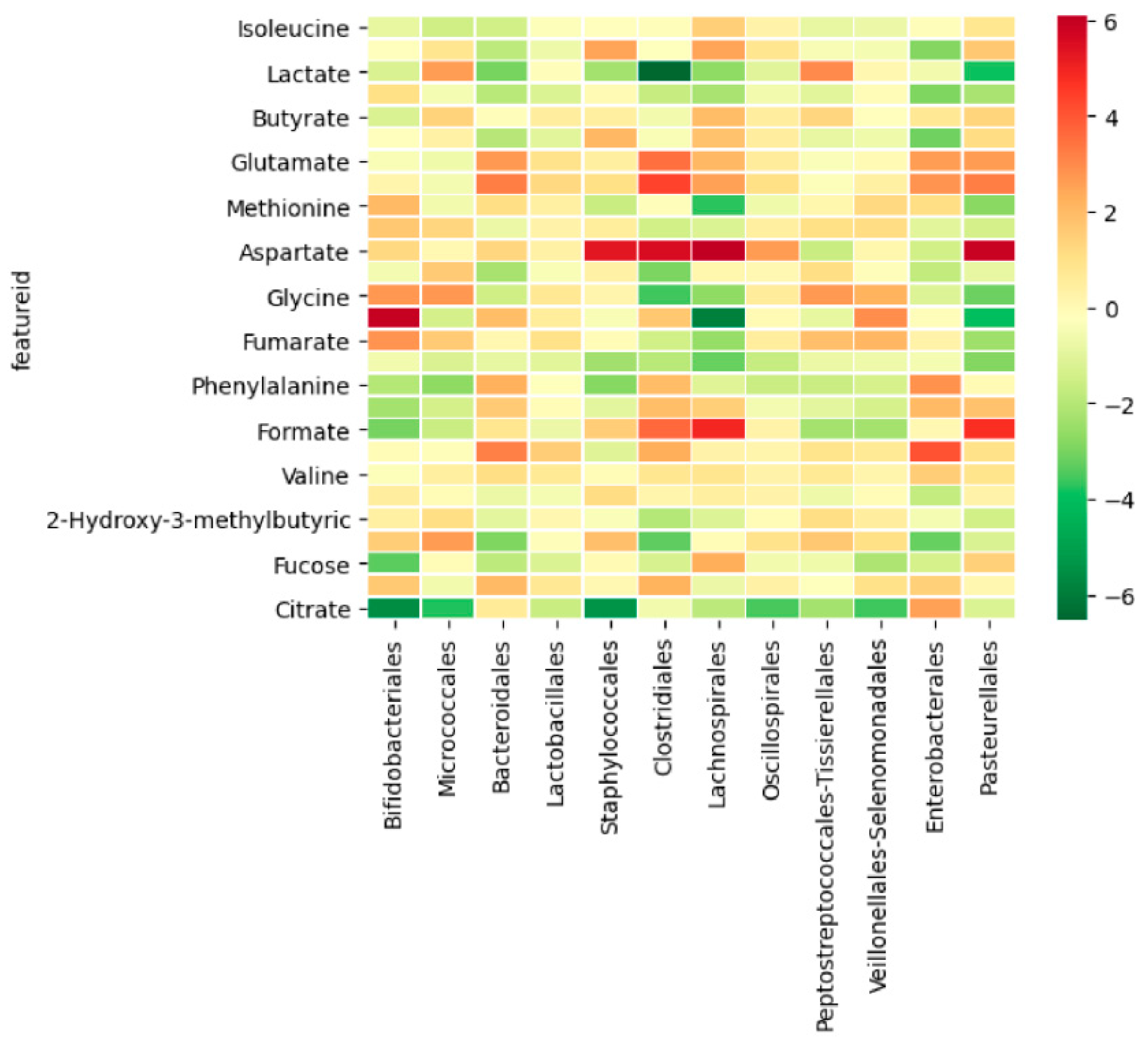
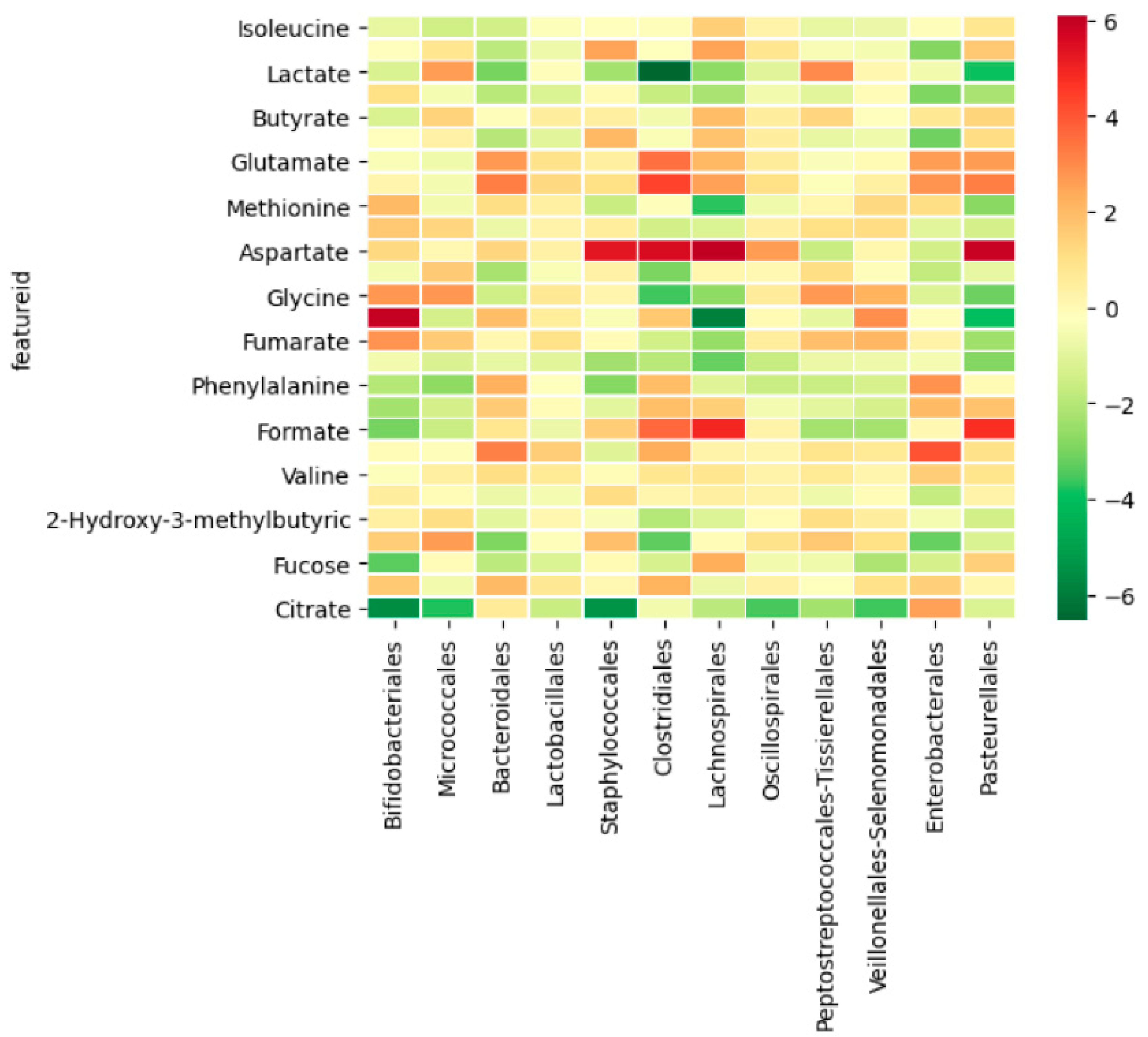
3.2.3. Integration between Metabolites and Gut Microbiome
4. Discussion
4.1. Method Optimization
4.2. Method Validation through a Multiomics Approach
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, T.; Santisteban, M.; Rodriguez, V.; Vermali, R.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Lin, H. Dysbiosis in gastrointestinal disorders. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 3–15. [Google Scholar] [CrossRef]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A.; de Giorgio, R.; Catassi, C.; Fasano, A. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, N.K. A systematic review of the prevalence and risk factors of irritable bowel syndrome among medical students. Turk. J. Gastroenterol. 2016, 27, 10–16. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Tilocca, B.; Pieroni, L.; Soggiu, A.; Britti, D.; Bonizzi, L.; Roncada, P.; Greco, V. Gut–Brain Axis and Neurodegeneration: State-of-the-Art of Meta-Omics Sciences for Microbiota Characterization. Int. J. Mol. Sci. 2020, 21, 4045. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]
- Guirro, M.; Costa, A.; Gual-Grau, A.; Mayneris-Perxachs, J.; Torrell, H.; Herrero, P.; Canela, N.; Arola, L. Multi-omics approach to elucidate the gut microbiota activity: Metaproteomics and metagenomics connection. Electrophoresis 2018, 39, 1692–1701. [Google Scholar] [CrossRef]
- Handelsman, J. Magnetic resonance in Pb x Nb y O z -ceramics as a system containing. Microbiol. Mol. Biol. Rev. 2004, 68, 1477–1481. [Google Scholar] [CrossRef]
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses (reverse transcriptase/dideoxynudeotide). Evolution 1985, 82, 6955–6959. [Google Scholar]
- Shaufi, M.A.M.; Sieo, C.C.; Chong, C.W.; Gan, H.M.; Ho, Y.W. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015, 7, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, S.; Meehan, C.J.; Van Puyvelde, S.; De Block, T.; Maltha, J.; Palpouguini, L.; Tahita, M.; Tinto, H.; Jacobs, J.; Deborggraeve, S. Diagnosis of Bacterial Bloodstream Infections: A 16S Metagenomics Approach. PLoS Negl. Trop. Dis. 2016, 10, e0004470. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Wang, Q.; O’Sullivan, O.; Greene-Diniz, R.; Cole, J.R.; Ross, R.P.; O’Toole, P.W. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010, 38, e200. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef] [Green Version]
- Pfleiderer, A.; Lagier, J.-C.; Armougom, F.; Robert, C.; Vialettes, B.; Raoult, D. Culturomics identified 11 new bacterial species from a single anorexia nervosa stool sample. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1471–1481. [Google Scholar] [CrossRef]
- Ku, H.-J.; Lee, J.-H. Development of a novel long-range 16S rRNA universal primer set for metagenomic analysis of gastrointestinal microbiota in newborn infants. J. Microbiol. Biotechnol. 2014, 24, 812–822. [Google Scholar] [CrossRef]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef]
- Sperling, J.L.; Silva-Brandão, K.; Brandão, M.; Lloyd, V.; Dang, S.; Davis, C.; Sperling, F.; Magor, K. Comparison of bacterial 16S rRNA variable regions for microbiome surveys of ticks. Ticks Tick-Borne Dis. 2017, 8, 453–461. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.; Olsen, G.J.; Maidak, B.L.; McCaughey, M.J.; Overbeek, R.A.; Macke, T.J.; Marsh, T.L.; Woese, C.R. The ribosomal database project. Nucleic Acids Res. 1993, 21, 3021–3023. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2011, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.-S.; Cho, Y.-J.; Lee, K.; Yoon, S.-H.; Kim, M.; Na, H.; Park, S.-C.; Jeon, Y.S.; Lee, J.-H.; Yi, H.; et al. Introducing EzTaxon-e: A prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 2012, 62, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.R.; Hester, J.; Kalady, M.; Xiao, H.; Li, X.; Serre, D. A novel method for determining microflora composition using dynamic phylogenetic analysis of 16S ribosomal RNA deep sequencing data. Genomics 2011, 98, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Handelsman, J.; Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; Souza, M.D.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Cai, Y.; Liu, L.; Yu, F.; Farrell, M.L.; Mckendree, W.; Farmerie, W. ESPRIT: Estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009, 37, e76. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cai, Y.; Mai, V.; Farmerie, W.; Yu, F.; Li, J.; Goodison, S. Advanced computational algorithms for microbial community analysis using massive 16S rRNA sequence data. Nucleic Acids Res. 2010, 38, e205. [Google Scholar] [CrossRef]
- Miller, C.S.; Baker, B.J.; Thomas, B.C.; Singer, S.W.; Banfield, J.F. EMIRGE: Reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011, 12, R44. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.H.; Kukkillaya, V.U.; Wilm, A.; Lay, C.; Ho, E.X.P.; Low, L.; Hibberd, M.L.; Nagarajan, N. Species Identification and Profiling of Complex Microbial Communities Using Shotgun Illumina Sequencing of 16S rRNA Amplicon Sequences. PLoS ONE 2013, 8, e60811. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2011, 13, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Hevia-Gonzalez, A.; Foroni, E.; Duranti, S.; Turroni, F.; Lugli, G.A.; Sanchez, B.; Martin, R.; Gueimonde, M.; Van Sinderen, D.; et al. Assessing the Fecal Microbiota: An Optimized Ion Torrent 16S rRNA Gene-Based Analysis Protocol. PLoS ONE 2013, 8, e68739. [Google Scholar] [CrossRef] [PubMed]
- Tridico, S.R.; Murray, D.C.; Addison, J.; Kirkbride, K.P.; Bunce, M. Metagenomic analyses of bacteria on human hairs: A qualitative assessment for applications in forensic science. Investig. Genet. 2014, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteley, A.; Jenkins, S.; Waite, I.; Kresoje, N.; Payne, H.; Mullan, B.; Allcock, R.; O’Donnell, A. Microbial 16S rRNA Ion Tag and community metagenome sequencing using the Ion Torrent (PGM) Platform. J. Microbiol. Methods 2012, 91, 80–88. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.Y. Conservative Fragments in Bacterial 16S rRNA Genes and Primer Design for 16S Ribosomal DNA Amplicons in Metagenomic Studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Edgar, R.C. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 2018, 34, 2371–2375. [Google Scholar] [CrossRef]
- Morton, J.T.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Van Goethem, M.; Northen, T.R.; Vazquez-Baeza, Y.; et al. Learning representations of microbe–metabolite interactions. Nat. Methods 2019, 16, 1306–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Parenti, M.; Grip, T.; Lönnerdal, B.; Timby, N.; Domellöf, M.; Hernell, O.; Slupsky, C.M. Fecal microbiome and metabolome of infants fed bovine MFGM supplemented formula or standard formula with breast-fed infants as reference: A randomized controlled trial. Sci. Rep. 2019, 9, 11589. [Google Scholar] [CrossRef] [Green Version]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Onywera, H.; Meiring, T.L. Comparative analyses of Ion Torrent V4 and Illumina V3-V4 16S rRNA gene metabarcoding methods for characterization of cervical microbiota: Taxonomic and functional profiling. Sci. Afr. 2020, 7, e00278. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, S.M.; Dethlefsen, L.; Huber, J.; Welch, D.M.; Relman, D.; Sogin, M.L. Exploring Microbial Diversity and Taxonomy Using SSU rRNA Hypervariable Tag Sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Liu, Z.; Lozupone, C.; Hamady, M.; Bushman, F.; Knight, R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007, 35, e120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soergel, D.A.W.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Segni, A.; Braun, T.; Benshoshan, M.; Barhom, S.F.; Saar, E.G.; Cesarkas, K.; Squires, J.E.; Keller, N.; Haberman, Y. Guided Protocol for Fecal Microbial Characterization by 16S rRNA-Amplicon Sequencing. J. Vis. Exp. 2018, 2018, e56845. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Duranti, S.; Viappiani, A.; Mangifesta, M.; Segata, N.; Van Sinderen, D.; Ventura, M. Evaluation of bifidobacterial community composition in the human gut by means of a targeted amplicon sequencing (ITS) protocol. FEMS Microbiol. Ecol. 2014, 90, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Caruso, V.; Song, X.; Asquith, M.; Karstens, L. Performance of Microbiome Sequence Inference Methods in Environments with Varying Biomass. mSystems 2019, 4, e00163-18. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Gregory Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Neupane, A.; Vo, R.; White, J.; Wang, X.; Marzano, S.-Y.L. Comparing Gut Microbiome in Mothers’ Own Breast Milk- and Formula-Fed Moderate-Late Preterm Infants. Front. Microbiol. 2020, 11, 891. [Google Scholar] [CrossRef]
- Ilcol, Y.O.; Ozbek, R.; Hamurtekin, E.; Ulus, I.H. Choline status in newborns, infants, children, breast-feeding women, breast-fed infants and human breast milk. J. Nutr. Biochem. 2005, 16, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo-Buitrago, M.E.; Sabater-Masdeu, M.; Moreno-Navarrete, J.M.; Infantes, E.C.; Rodríguez, M.A.; Coll, C.; Ramió, L.; Palomino-Schätzlein, M.; Gutiérrez-Carcedo, P.; Pérez-Brocal, V.; et al. Glutamate interactions with obesity, insulin resistance, cognition and gut microbiota composition. Acta Diabetol. 2019, 56, 569–579. [Google Scholar] [CrossRef]
- Salazar, N.; Dewulf, E.M.; Neyrinck, A.; Bindels, L.B.; Cani, P.D.; Mahillon, J.; de Vos, W.M.; Thissen, J.-P.; Gueimonde, M.; Reyes-Gavilan, C.D.L.; et al. Inulin-type fructans modulate intestinal Bifidobacterium species populations and decrease fecal short-chain fatty acids in obese women. Clin. Nutr. 2015, 34, 501–507. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name and Direction | Adaptor | Key | Barcode | Spacer | Target 16S Primer |
|---|---|---|---|---|---|
| V3_forward | CCATCTCATCCCTGCGTGTCTCCGAC | TCAG | CTAAGGTAAC | GAT | CCTACGGGRSGCAGCAG |
| V3_reverse | CCTCTCTATGGGCAGTCGGTGAT | CC | ATTACCGCGGCTGCT | ||
| V4_forward | CCATCTCATCCCTGCGTGTCTCCGAC | TCAG | CTAAGGTAAC | GAT | GTGCCAGCMGCCGCGGTAA |
| V4_reverse | CCTCTCTATGGGCAGTCGGTGAT | CC | GGACTACHVGGGTWTCTAAT | ||
| V6_forward | CCATCTCATCCCTGCGTGTCTCCGAC | TCAG | CTAAGGTAAC | GAT | CAACGCGAAGAACCTTACC |
| V6_reverse | CCTCTCTATGGGCAGTCGGTGAT | CC | CGACAGCCATGCANCACCT |
| Library Construction | Feature Selection | Taxonomy | Bray–Curtis Dissimilarity | ||
|---|---|---|---|---|---|
| Species | Genus | Family | |||
| V4 | ASV | VSEARCH | 0.5576 | 0.1182 | 0.1182 |
| V3_V4 | ASV | VSEARCH | 0.5577 | 0.1182 | 0.1182 |
| V3 | ASV | VSEARCH | 0.5930 | 0.1186 | 0.1186 |
| V3_V4_V6 | ASV | VSEARCH | 0.6221 | 0.2084 | 0.1756 |
| V4_V6 | ASV | VSEARCH | 0.6222 | 0.2087 | 0.1758 |
| V3 | OTU | VSEARCH | 0.6966 | 0.2342 | 0.1405 |
| V3_V6 | OTU | VSEARCH | 0.7268 | 0.2617 | 0.1903 |
| V3_V6 | OTU | BLAST | 0.7627 | 0.2601 | 0.1916 |
| V3 | OTU | sklearn | 0.7695 | 0.2387 | 0.1445 |
| kit | OTU | VSEARCH | 0.7712 | 0.2506 | 0.1532 |
| V6 | OTU | BLAST | 0.7770 | 0.3955 | 0.3580 |
| V3_V4 | OTU | VSEARCH | 0.7791 | 0.4680 | 0.4374 |
| kit | ASV | BLAST | 0.7792 | 0.3138 | 0.3137 |
| V3_V4_V6 | OTU | VSEARCH | 0.7825 | 0.4392 | 0.4086 |
| V3 | OTU | BLAST | 0.7872 | 0.2338 | 0.1405 |
| V6 | OTU | VSEARCH | 0.7916 | 0.3961 | 0.3562 |
| kit | ASV | VSEARCH | 0.7978 | 0.3138 | 0.3137 |
| kit | OTU | BLAST | 0.8077 | 0.2477 | 0.1529 |
| V3_V4_V6 | OTU | BLAST | 0.8088 | 0.4386 | 0.4096 |
| V3_V4 | OTU | BLAST | 0.8208 | 0.4682 | 0.4378 |
| V4_V6 | OTU | VSEARCH | 0.8262 | 0.5376 | 0.5264 |
| V4 | ASV | BLAST | 0.8305 | 0.1182 | 0.1182 |
| V3_V4 | ASV | BLAST | 0.8307 | 0.1182 | 0.1182 |
| V4_V6 | OTU | BLAST | 0.8312 | 0.5371 | 0.5278 |
| V4_V6 | ASV | BLAST | 0.8351 | 0.2088 | 0.1759 |
| V3_V4_V6 | ASV | BLAST | 0.8351 | 0.2085 | 0.1757 |
| V3_V6 | ASV | VSEARCH | 0.8413 | 0.4719 | 0.4536 |
| V6 | ASV | VSEARCH | 0.8438 | 0.4761 | 0.4577 |
| V6 | ASV | BLAST | 0.8443 | 0.5015 | 0.4584 |
| V3_V6 | ASV | BLAST | 0.8445 | 0.4970 | 0.4543 |
| V4 | OTU | VSEARCH | 0.8454 | 0.6350 | 0.6346 |
| kit | OTU | sklearn | 0.8568 | 0.3560 | 0.2626 |
| V4 | OTU | BLAST | 0.8614 | 0.6355 | 0.6353 |
| V3 | ASV | BLAST | 0.8631 | 0.1186 | 0.1186 |
| kit | ASV | sklearn | 0.8724 | 0.5185 | 0.5181 |
| V3_V6 | OTU | sklearn | 0.8756 | 0.3614 | 0.2275 |
| V3 | ASV | sklearn | 0.8914 | 0.1186 | 0.1186 |
| V3_V4 | ASV | sklearn | 0.8972 | 0.1182 | 0.1182 |
| V4 | ASV | sklearn | 0.8973 | 0.1182 | 0.1182 |
| V3_V4 | OTU | sklearn | 0.9144 | 0.6126 | 0.5830 |
| V3_V4_V6 | ASV | sklearn | 0.9282 | 0.2321 | 0.1864 |
| V4_V6 | ASV | sklearn | 0.9284 | 0.2325 | 0.1869 |
| V3_V4_V6 | OTU | sklearn | 0.9367 | 0.5926 | 0.5302 |
| V3_V6 | ASV | sklearn | 0.9958 | 0.6680 | 0.5621 |
| V6 | OTU | sklearn | 0.9958 | 0.5720 | 0.4090 |
| V6 | ASV | sklearn | 0.9968 | 0.6740 | 0.5672 |
| V4_V6 | OTU | sklearn | 0.9985 | 0.7347 | 0.6812 |
| V4 | OTU | sklearn | 1.0000 | 0.8434 | 0.8434 |
| Species | Expected | Obtained V3 + V4 | Obtained V3 | Obtained V4 |
|---|---|---|---|---|
| Lactobacillus brevis | 9.09% | 8.80% | 6.62% | 8.80% |
| Pediococcus pentosaceus | 9.09% | 10.05% | 8.83% | 10.06% |
| Lactobacillus plantarum | 9.09% | 11.89% | 11.48% | 11.89% |
| Gluconobacter oxydans | 9.09% | 8.14% | 7.06% | 8.14% |
| Bacteroides coprophilus | 9.09% | 0.00% | 0.00% | 0.00% |
| Lactobacillus hilgardii | 9.09% | 0.02% | 0.00% | 0.02% |
| Escherichia coli | 9.09% | 9.99% | 9.93% | 9.99% |
| Prevotella copri | 9.09% | 0.00% | 0.00% | 0.00% |
| Pediococcus parvulus | 9.09% | 0.00% | 0.00% | 0.00% |
| Acetobacter malorum | 9.09% | 0.00% | 0.00% | 0.00% |
| Lactobacillus buchneri | 9.09% | 0.00% | 0.00% | 0.00% |
| Other | 0 | 51.11% | 56.07% | 51.09% |
| Genus | Expected | Obtained V3 + V4 | Obtained V3 | Obtained V4 |
| Bacteroides | 9.09% | 9.33% | 10.30% | 9.32% |
| Prevotella | 9.09% | 7.63% | 8.68% | 7.62% |
| Lactobacillus | 36.36% | 37.08% | 36.72% | 37.08% |
| Pediococcus | 18.18% | 18.96% | 18.54% | 18.96% |
| Acetobacter | 9.09% | 8.76% | 8.76% | 8.76% |
| Gluconobacter | 9.09% | 8.14% | 7.06% | 8.15% |
| Escherichia | 9.09% | 9.99% | 9.93% | 9.99% |
| Other | 0.00% | 0.10% | 0.00% | 0.10% |
| Family | Expected | Obtained V3 + V4 | Obtained V3 | Obtained V4 |
| Bacteroidaceae | 9.09% | 9.33% | 10.30% | 9.32% |
| Prevotellaceae | 9.09% | 7.63% | 8.68% | 7.62% |
| Lactobacillaceae | 54.55% | 56.04% | 55.26% | 56.04% |
| Acetobacteraceae | 18.18% | 16.90% | 15.82% | 16.91% |
| Enterobacteriaceae | 9.09% | 9.99% | 9.93% | 9.99% |
| Other | 0 | 0.10% | 0.00% | 0.10% |
| Breast-Fed Group | Formula-Fed Group | |||||
|---|---|---|---|---|---|---|
| Metabolite | Mean | Desvest | SEM | Mean | Desvest | SEM |
| Valine | 1.25 | 0.76 | 0.25 | 3.51 | 2.43 | 0.99 |
| Uracil | 0.05 | 0.06 | 0.02 | 0.14 | 0.08 | 0.04 |
| Tyrosine | 0.12 | 0.08 | 0.03 | 0.16 | 0.09 | 0.04 |
| Tryprophan | 0.06 | 0.05 | 0.02 | 0.11 | 0.06 | 0.02 |
| Succinate | 2.12 | 2.12 | 0.71 | 6.17 | 10.47 | 4.28 |
| Propionate | 1.73 | 1.24 | 0.41 | 2.33 | 1.23 | 0.50 |
| Phenylalanine | 0.12 | 0.07 | 0.02 | 0.30 | 0.17 | 0.07 |
| Methionine sulfoxide | 0.03 | 0.03 | 0.01 | 0.04 | 0.04 | 0.02 |
| Methionine | 0.14 | 0.08 | 0.03 | 0.33 | 0.20 | 0.08 |
| Leucine | 0.69 | 0.39 | 0.13 | 1.58 | 1.05 | 0.43 |
| Lactate | 0.48 | 0.37 | 0.12 | 7.82 | 8.12 | 3.32 |
| Isoleucine | 0.79 | 0.51 | 0.17 | 1.96 | 1.17 | 0.48 |
| Hypoxanthine | 0.06 | 0.03 | 0.01 | 0.10 | 0.07 | 0.03 |
| Glycine | 0.41 | 0.18 | 0.06 | 2.61 | 2.17 | 0.89 |
| Glutamate | 0.83 | 0.71 | 0.24 | 1.48 | 1.08 | 0.44 |
| Fumarate | 0.03 | 0.02 | 0.01 | 0.02 | 0.01 | 0.00 |
| Fucose | 0.21 | 0.30 | 0.10 | 0.25 | 0.17 | 0.07 |
| Formate | 0.02 | 0.01 | 0.00 | 0.07 | 0.07 | 0.03 |
| Citrate | 0.18 | 0.15 | 0.05 | 1.18 | 1.15 | 0.47 |
| Choline | 0.07 | 0.04 | 0.01 | 0.17 | 0.06 | 0.02 |
| Butyrate | 1.21 | 1.24 | 0.41 | 3.99 | 2.78 | 1.13 |
| Aspartate | 0.21 | 0.12 | 0.04 | 0.67 | 0.70 | 0.29 |
| Alanine | 1.31 | 0.86 | 0.29 | 3.24 | 2.06 | 0.84 |
| Acetate | 6.72 | 5.71 | 1.90 | 16.64 | 12.19 | 4.98 |
| 5-Aminopentanoate | 0.82 | 1.70 | 0.57 | 1.22 | 1.41 | 0.58 |
| 2-Hydroxy-3-methylbutyric | 0.04 | 0.03 | 0.01 | 0.11 | 0.07 | 0.03 |
| 2-3-Butanediol | 0.23 | 0.16 | 0.05 | 0.44 | 0.60 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrell, H.; Cereto-Massagué, A.; Kazakova, P.; García, L.; Palacios, H.; Canela, N. Multiomic Approach to Analyze Infant Gut Microbiota: Experimental and Analytical Method Optimization. Biomolecules 2021, 11, 999. https://doi.org/10.3390/biom11070999
Torrell H, Cereto-Massagué A, Kazakova P, García L, Palacios H, Canela N. Multiomic Approach to Analyze Infant Gut Microbiota: Experimental and Analytical Method Optimization. Biomolecules. 2021; 11(7):999. https://doi.org/10.3390/biom11070999
Chicago/Turabian StyleTorrell, Helena, Adrià Cereto-Massagué, Polina Kazakova, Lorena García, Héctor Palacios, and Núria Canela. 2021. "Multiomic Approach to Analyze Infant Gut Microbiota: Experimental and Analytical Method Optimization" Biomolecules 11, no. 7: 999. https://doi.org/10.3390/biom11070999
APA StyleTorrell, H., Cereto-Massagué, A., Kazakova, P., García, L., Palacios, H., & Canela, N. (2021). Multiomic Approach to Analyze Infant Gut Microbiota: Experimental and Analytical Method Optimization. Biomolecules, 11(7), 999. https://doi.org/10.3390/biom11070999