
Synuclein Family Members Prevent Membrane Damage by Counteracting α-Synuclein Aggregation

 , and
, and 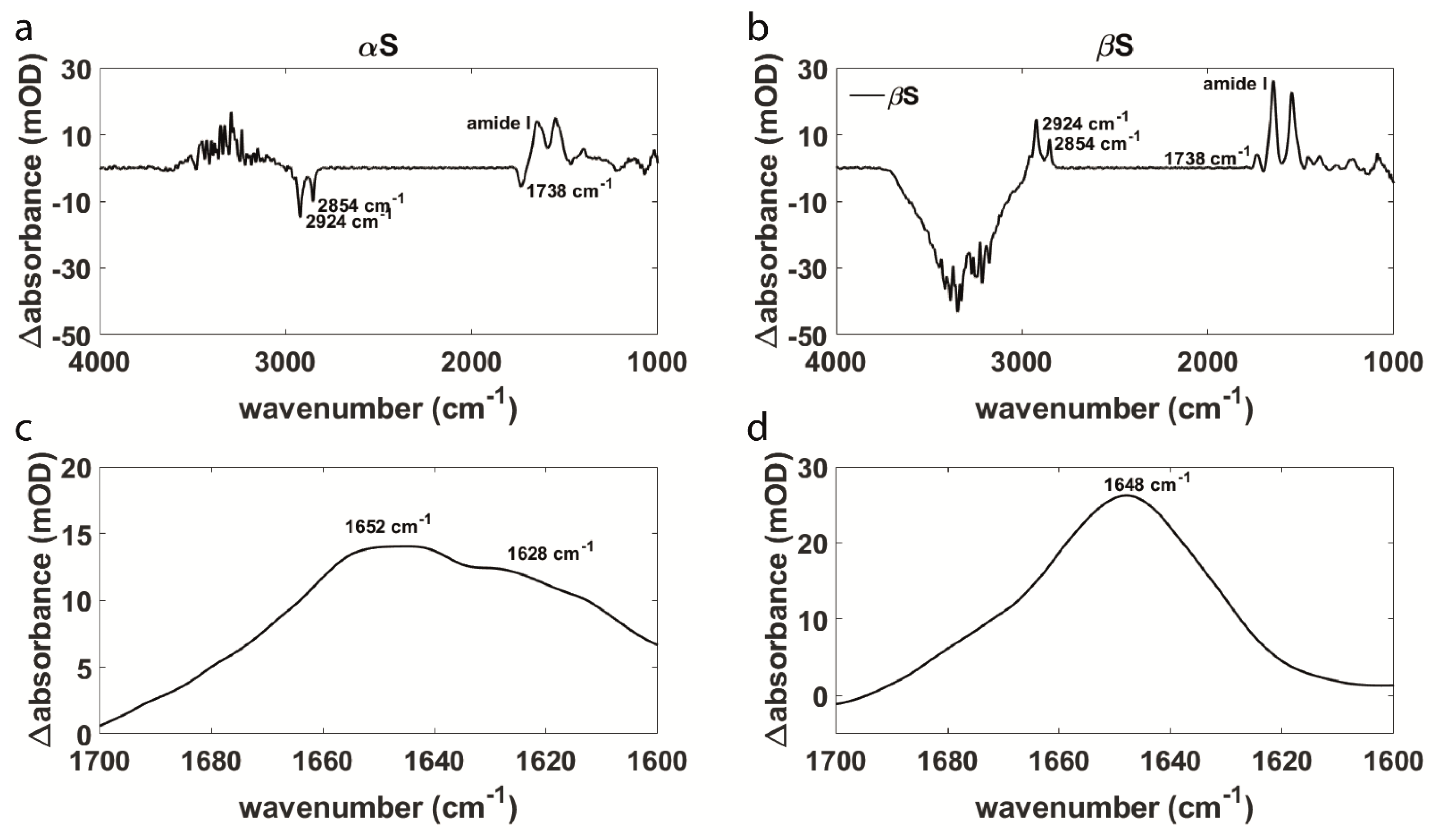{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Biomimetic Membranes
2.3. Sample Preparation
2.4. Polarized ATR-FTIR
3. Results & Discussion
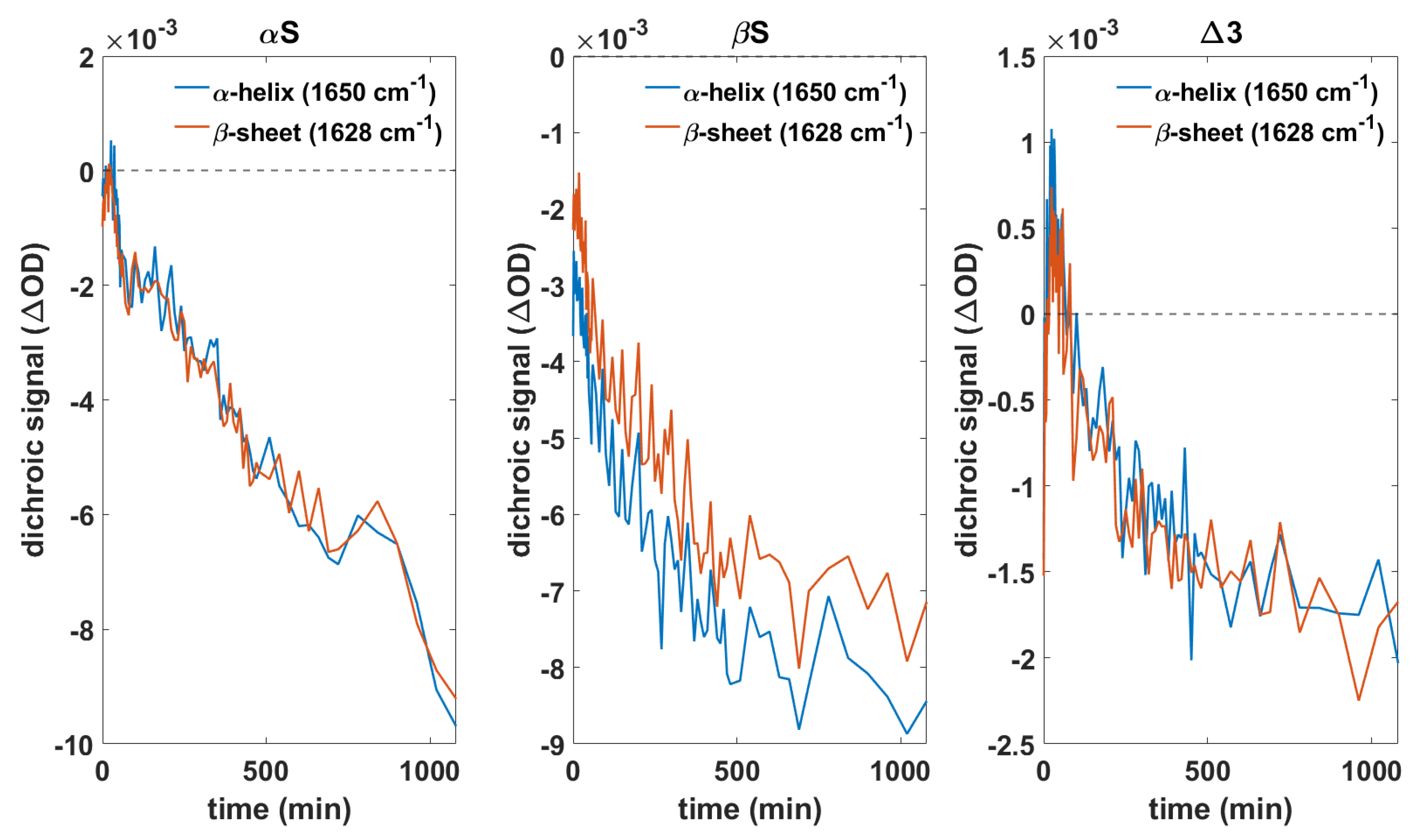
3.1. Aggregation Behavior and Membrane Interaction Are Significantly Different for α- and β-Synuclein
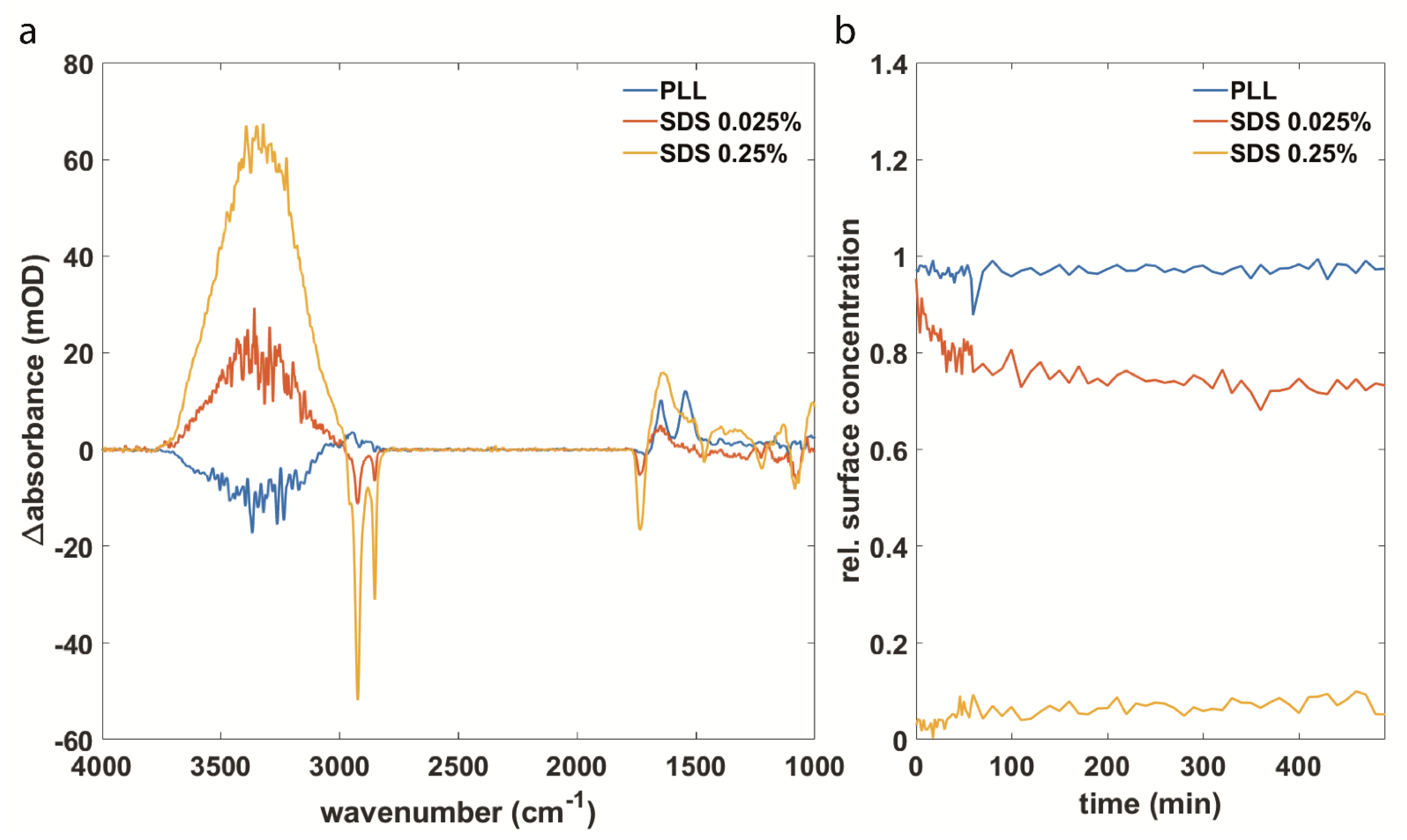
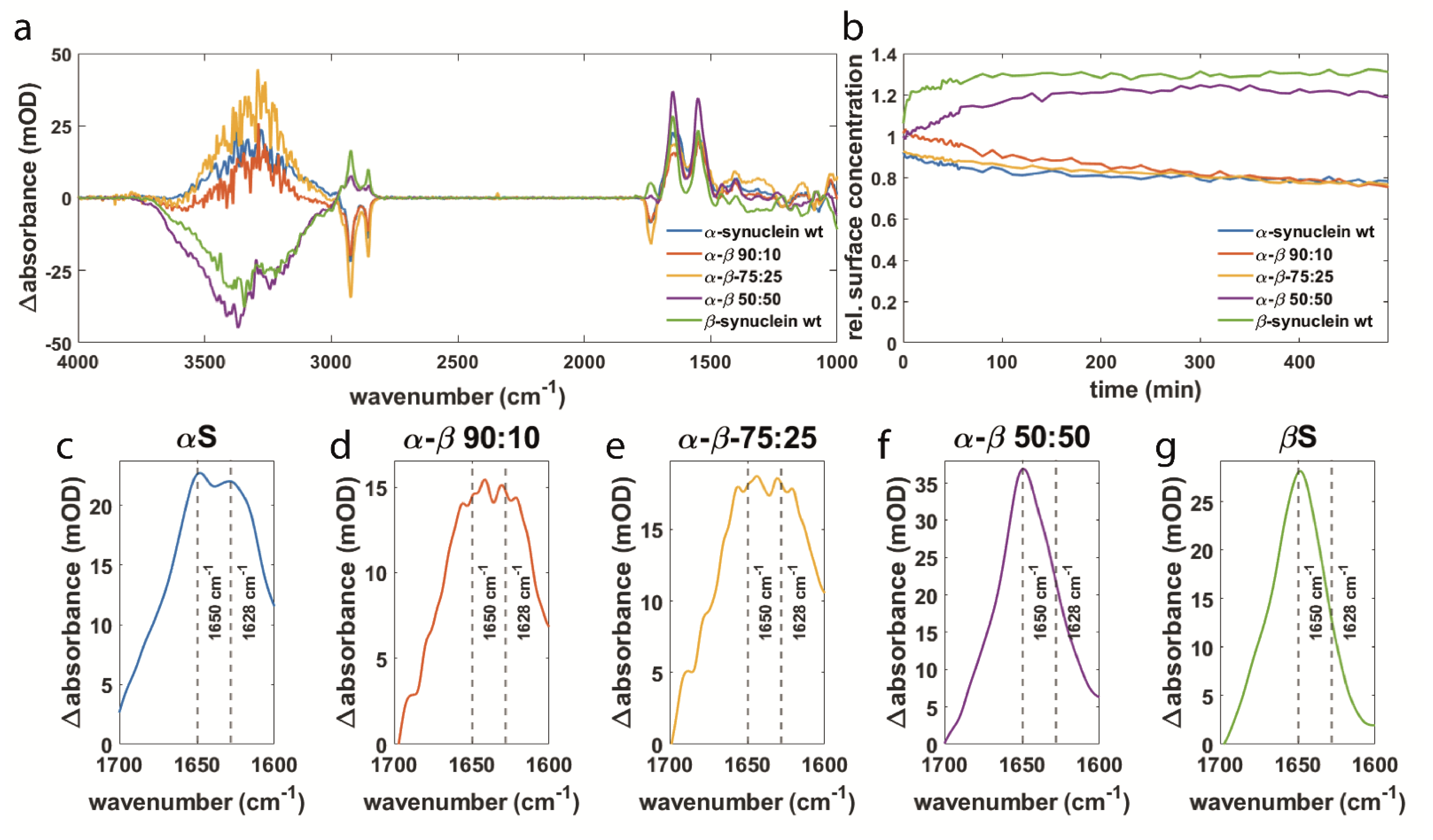
3.2. Synuclein Interaction Prevents α-Synuclein Aggregation and Maintains Membrane Integrity
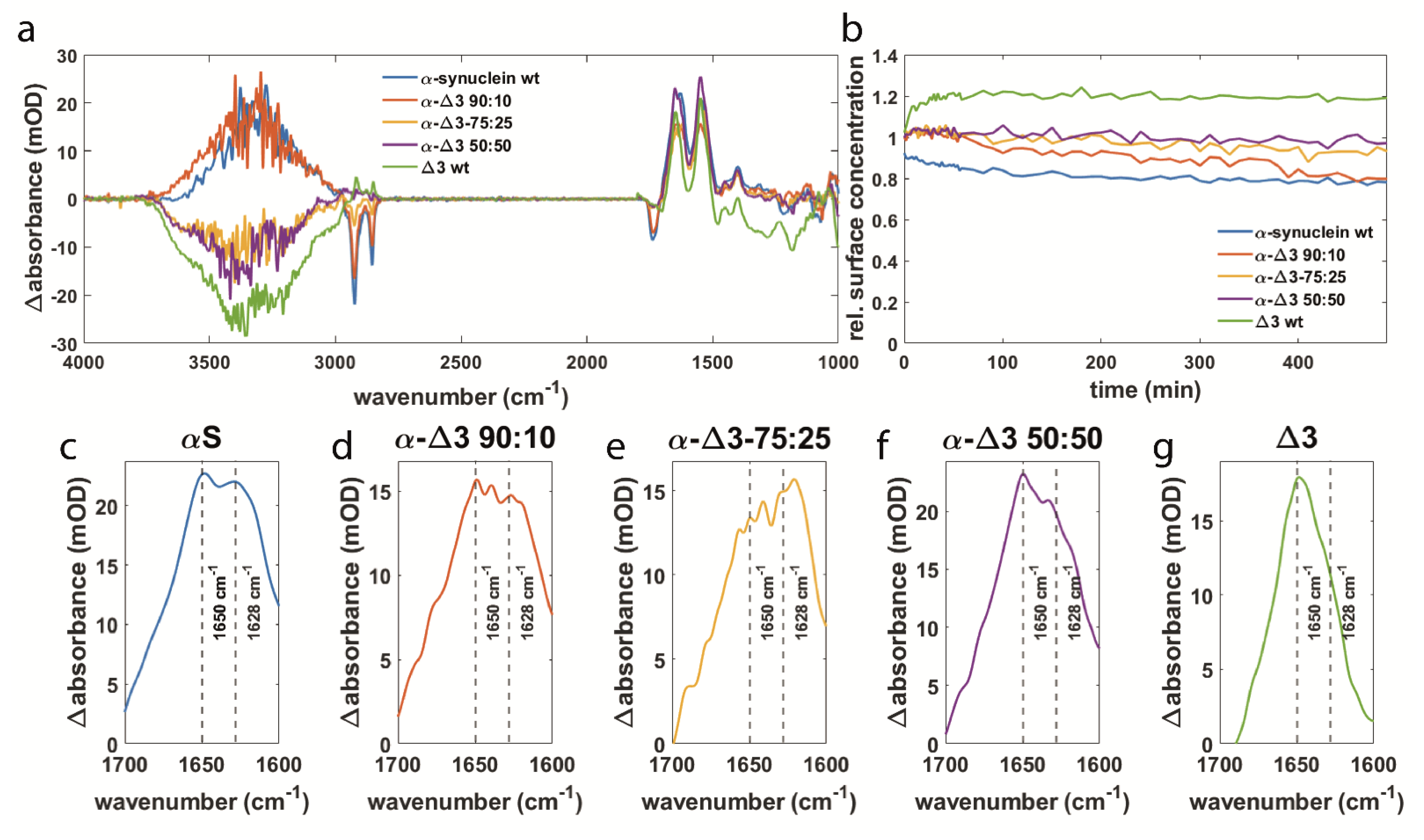
3.3. αS Δexon3 Shows Similar Effects to β-Synuclein but to a Lesser Extent
3.4. Orientation of Secondary Structure Elements upon Membrane Interaction Studied by Polarized Measurements
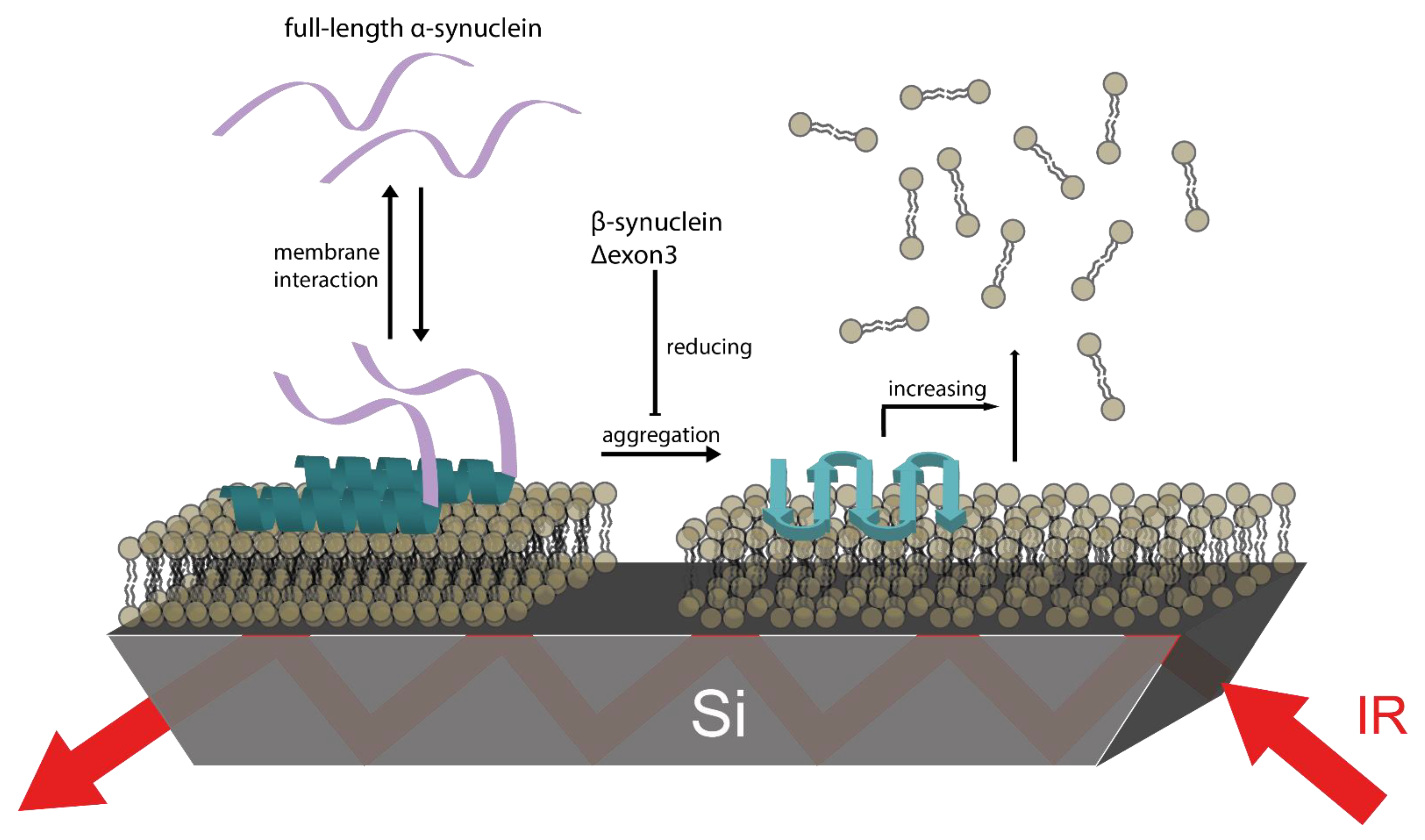
3.5. βS and Δ3 Prevent Membrane Damage by Counteracting αS Aggregation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Lou, X.; Kim, J.; Hawk, B.J.; Shin, Y.-K. α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 2017, 474, 2039–2049. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. α-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Diao, J.; Burré, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Südhof, T.C.; Brunger, A.T. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. ELife 2013, 2, e00592. [Google Scholar] [CrossRef]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Caughey, B.; Peter, T.; Lansbury, J. PROTOFIBRILS, PORES, FIBRILS, AND NEURODEGENERATION: Separating the Responsible Protein Aggregates from The Innocent Bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Van Rooijen, B.D.; Claessens, M.M.; Subramaniam, V. Membrane Permeabilization by Oligomeric alpha-Synuclein: In Search of the Mechanism. PLoS ONE 2010, 5, e14292. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.L.; Brown, D.R. Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef]
- Sciacca, M.F.; Lolicato, F.; Tempra, C.; Scollo, F.; Sahoo, B.R.; Watson, M.D.; Garcia-Vinuales, S.; Milardi, D.; Raudino, A.; Lee, J.C.; et al. Lipid-Chaperone Hypothesis: A Common Molecular Mechanism of Membrane Disruption by Intrinsically Disordered Proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [Google Scholar] [CrossRef]
- Afitska, K.; Fucikova, A.; Shvadchak, V.V.; Yushchenko, D.A. α-Synuclein aggregation at low concentrations. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 701–709. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. α-Synuclein Binds Large Unilamellar Vesicles as an Extended Helix. Biochemistry 2009, 48, 2304–2306. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-bound Human α-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef]
- Fallah, M.A.; Gerding, H.R.; Scheibe, C.; Drescher, M.; Karreman, C.; Schildknecht, S.; Leist, M.; Hauser, K. Simultaneous IR-Spectroscopic Observation of alpha-Synuclein, Lipids, and Solvent Reveals an Alternative Membrane-Induced Oligomerization Pathway. ChemBiochem A Eur. J. Chem. Biol. 2017, 18, 2312–2316. [Google Scholar] [CrossRef]
- Bisaglia, M.; Tessari, I.; Mammi, S.; Bubacco, L. Interaction Between alpha-Synuclein and Metal Ions, Still Looking for a Role in the Pathogenesis of Parkinson’s Disease. Neuromol. Med. 2009, 11, 239–251. [Google Scholar] [CrossRef]
- Lowe, R.; Pountney, D.L.; Jensen, P.H.; Gai, W.P.; Voelcker, N.H. Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004, 13, 3245–3252. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhang, Z.T.; Liu, M.L.; Li, C.G. Alpha-synuclein-lanthanide metal ions interaction: Binding sites, conformation and fibrillation. BMC Biophys. 2016, 9, 1. [Google Scholar] [CrossRef]
- Kozlowski, H.; Luczkowski, M.; Remelli, M.; Valensin, D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. 2012, 256, 2129–2141. [Google Scholar] [CrossRef]
- Madine, J.; Doig, A.J.; Middleton, D.A. A Study of the Regional Effects of α-Synuclein on the Organization and Stability of Phospholipid Bilayers. Biochemistry 2006, 45, 5783–5792. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Proukakis, C.; Houlden, H.; Schapira, A.H. Somatic alpha-synuclein mutations in Parkinson’s disease: Hypothesis and preliminary data. Mov. Disord. 2013, 28, 705–712. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian–pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Fujioka, S.; Ogaki, K.; Tacik, P.M.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat. Disord. 2014, 20, S29–S34. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef]
- Ruf, V.C.; Nübling, G.S.; Willikens, S.; Shi, S.; Schmidt, F.; Levin, J.; Bötzel, K.; Kamp, F.; Giese, A. Different Effects of α-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. ACS Chem. Neurosci. 2019, 10, 1649–1659. [Google Scholar] [CrossRef]
- Campion, D.; Martin, C.; Heilig, R.; Charbonnier, F.; Moreau, V.; Flaman, J.M.; Petit, J.L.; Hannequin, D.; Brice, A.; Frebourg, T. The NACP/synuclein gene: Chromosomal assignment and screening for alterations in Alzheimer disease. Genomics 1995, 26, 254–257. [Google Scholar] [CrossRef]
- Beyer, K.; Humbert, J.; Ferrer, A.; Lao, J.I.; Carrato, C.; López, D.; Ferrer, I.; Ariza, A. Low alpha-synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. NeuroReport 2006, 17, 1327–1330. [Google Scholar] [CrossRef]
- Ducas, V.C.; Rhoades, E. Quantifying Interactions of β-Synuclein and γ-Synuclein with Model Membranes. J. Mol. Biol. 2012, 423, 528–539. [Google Scholar] [CrossRef]
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Kröhnert, K.; Schäfer, C.; Rammner, B.; Koo, S.J.; Claßen, G.A.; Krauss, M.; Haucke, V.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028. [Google Scholar] [CrossRef]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Iljina, M. Oligomerisation of Alpha-Synuclein at Physiological Concentrations. Biophys. J. 2014, 106, 267A–268A. [Google Scholar] [CrossRef][Green Version]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical Properties of the Synucleins and Their Propensities to Fibrillate: INHIBITION OF α-SYNUCLEIN ASSEMBLY BY β- AND γ-SYNUCLEINS*. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef]
- Moriarty, G.M.; Olson, M.P.; Atieh, T.B.; Janowska, M.K.; Khare, S.D.; Baum, J. A pH-dependent switch promotes β-synuclein fibril formation via glutamate residues. J. Biol. Chem. 2017, 292, 16368–16379. [Google Scholar] [CrossRef]
- Yamin, G.; Munishkina, L.A.; Karymov, M.A.; Lyubchenko, Y.L.; Uversky, V.N.; Fink, A.L. Forcing Nonamyloidogenic β-Synuclein To Fibrillate. Biochemistry 2005, 44, 9096–9107. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.P.; Meisl, G.; Knowles, T.P.J.; Buell, A.K.; Dobson, C.M.; Galvagnion, C. Kinetic barriers to α-synuclein protofilament formation and conversion into mature fibrils. Chem. Commun. 2018, 54, 7854–7857. [Google Scholar] [CrossRef]
- Ohtake, H.; Limprasert, P.; Fan, Y.; Onodera, O.; Kakita, A.; Takahashi, H.; Bonner, L.T.; Tsuang, D.W.; Murray, I.V.J.; Lee, V.M.-Y.; et al. β-Synuclein gene alterations in dementia with Lewy bodies. Neurology 2004, 63, 805–811. [Google Scholar] [CrossRef]
- Wright, J.A.; McHugh, P.C.; Pan, S.; Cunningham, A.; Brown, D.R. Counter-regulation of alpha- and beta-synuclein expression at the transcriptional level. Mol. Cell. Neurosci. 2013, 57, 33–41. [Google Scholar] [CrossRef]
- Fink, A.L. The aggregation and fibrillation of alpha-synuclein. Acc. Chem. Res. 2006, 39, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.P.; Buell, A.K.; Michaels, T.C.T.; Meisl, G.; Carozza, J.; Flagmeier, P.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Galvagnion, C. Beta-Synuclein suppresses both the initiation and amplification steps of alpha-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 2016, 6, 36010. [Google Scholar] [CrossRef]
- Janowska, M.K.; Wu, K.-P.; Baum, J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci. Rep. 2015, 5, 15164. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Crews, L.; Bar-On, P.; Gage, F.H.; Marr, R.; Masliah, E. An antiaggregation gene therapy strategy for Lewy body disease utilizing β-synuclein lentivirus in a transgenic model. Gene Ther. 2004, 11, 1713–1723. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. β-Synuclein Inhibits α-Synuclein Aggregation: A Possible Role as an Anti-Parkinsonian Factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef]
- Windisch, M.; Hutter-Paier, B.; Rockenstein, E.; Hashimoto, M.; Mallory, M.; Masliah, E. Development of a new treatment for Alzheimer’s disease and parkinson’s disease using anti-aggregatory β-synuclein-derived peptides. J. Mol. Neurosci. 2002, 19, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Yang, X.; Baum, J. Interactions between the Intrinsically Disordered Proteins beta-Synuclein and alpha-Synuclein. Proteomics 2018, 18, 1800109. [Google Scholar] [CrossRef] [PubMed]
- Taschenberger, G.; Toloe, J.; Tereshchenko, J.; Akerboom, J.; Wales, P.; Benz, R.; Becker, S.; Outeiro, T.F.; Looger, L.L.; Bähr, M.; et al. β-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann. Neurol. 2013, 74, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef]
- Beyer, K.; Ariza, A. Alpha-Synuclein Posttranslational Modification and Alternative Splicing as a Trigger for Neurodegeneration. Mol. Neurobiol. 2013, 47, 509–524. [Google Scholar] [CrossRef]
- Bungeroth, M.; Appenzeller, S.; Regulin, A.; Völker, W.; Lorenzen, I.; Grötzinger, J.; Pendziwiat, M.; Kuhlenbäumer, G. Differential aggregation properties of alpha-synuclein isoforms. Neurobiol. Aging 2014, 35, 1913–1919. [Google Scholar] [CrossRef]
- Valastyan, J.S.; Termine, D.J.; Lindquist, S. Splice isoform and pharmacological studies reveal that sterol depletion relocalizes α-synuclein and enhances its toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 3014–3019. [Google Scholar] [CrossRef]
- Beyer, K.; Lao, J.I.; Carrato, C.; Mate, J.L.; López, D.; Ferrer, I.; Ariza, A. Differential expression of α-synuclein isoforms in dementia with Lewy bodies. Neuropathol. Appl. Neurobiol. 2004, 30, 601–607. [Google Scholar] [CrossRef]
- Muñoz, E.; Pastor, P.; Martí, M.J.; Valldeoriola, F.; Tolosa, E.; Oliva, R. Enfermedad de Parkinson esporádica y familiar: Estudio comparativo. Med. Clín. 2001, 116, 601–604. [Google Scholar] [CrossRef]
- Zhang, G.; Keiderling, T.A. Equilibrium and Dynamic Spectroscopic Studies of the Interaction of Monomeric β-Lactoglobulin with Lipid Vesicles at Low pH. Biochemistry 2014, 53, 3079–3087. [Google Scholar] [CrossRef]
- Scheibe, C.; Hauser, K. Orientation of lipids in solid supported lipid bilayers studied by polarized ATR-FTIR spectroscopy. Biomed. Spectrosc. Imaging 2018, 7, 17–24. [Google Scholar] [CrossRef]
- Klima, S.; Brull, M.; Spreng, A.S.; Suciu, I.; Falt, T.; Schwamborn, J.C.; Waldmann, T.; Karreman, C.; Leist, M. A human stem cell-derived test system for agents modifying neuronal N-methyl-D-aspartate-type glutamate receptor Ca(2+)-signalling. Arch. Toxicol. 2021, 95, 1703–1722. [Google Scholar] [CrossRef]
- Karreman, C. Fusion PCR, a one-step variant of the “megaprimer” method of mutagenesis. Biotechniques 1998, 24, 736–742. [Google Scholar] [CrossRef]
- Dubendorff, J.W.; Studier, F.W. Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol. 1991, 219, 45–59. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Raussens, V.; Ruysschaert, J.M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1999, 1422, 105–185. [Google Scholar] [CrossRef]
- Tatulian, S.A. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy: A Method of Choice for Studying Membrane Proteins and Lipids. Biochemistry 2003, 42, 11898–11907. [Google Scholar] [CrossRef]
- Hassler, N.; Baurecht, D.; Reiter, G.; Fringeli, U.P. In Situ FTIR ATR Spectroscopic Study of the Interaction of Immobilized Human Serum Albumin with Cholate in Aqueous Environment. J. Phys. Chem. C 2011, 115, 1064–1072. [Google Scholar] [CrossRef]
- Kötting, C.; Güldenhaupt, J.; Gerwert, K. Time-resolved FTIR spectroscopy for monitoring protein dynamics exemplified by functional studies of Ras protein bound to a lipid bilayer. Chem. Phys. 2012, 396, 72–83. [Google Scholar] [CrossRef]
- Tatulian, S.A.; Jones, L.R.; Reddy, L.G.; Stokes, D.L.; Tamm, L.K. Secondary structure and orientation of phospholamban reconstituted in supported bilayers from polarized attenuated total reflection FTIR spectroscopy. Biochemistry 1995, 34, 4448–4456. [Google Scholar] [CrossRef] [PubMed]
- Sarroukh, R.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. ATR-FTIR: A “rejuvenated” tool to investigate amyloid proteins. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2328–2338. [Google Scholar] [CrossRef]
- Trombik, P.; Cieślik-Boczula, K. Influence of phenothiazine molecules on the interactions between positively charged poly-l-lysine and negatively charged DPPC/DPPG membranes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 227, 117563. [Google Scholar] [CrossRef]
- Beyer, K. α-Synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T. α-Synuclein, Especially the Parkinson’s Disease-associated Mutants, Forms Pore-like Annular and Tubular Protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Schmidt, F.; Levin, J.; Kamp, F.; Kretzschmar, H.; Giese, A.; Botzel, K. Single-Channel Electrophysiology Reveals a Distinct and Uniform Pore Complex Formed by alpha-Synuclein Oligomers in Lipid Membranes. PLoS ONE 2012, 7, e42545. [Google Scholar] [CrossRef]
- Tosatto, L.; Andrighetti, A.O.; Plotegher, N.; Antonini, V.; Tessari, I.; Ricci, L.; Bubacco, L.; Serra, M.D. Alpha-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2876–2883. [Google Scholar] [CrossRef]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef]
- Ramalingam, N.; Dettmer, U. Temperature is a key determinant of alpha- and beta-synuclein membrane interactions in neurons. J. Biol. Chem. 2021, 296, 100271. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheibe, C.; Karreman, C.; Schildknecht, S.; Leist, M.; Hauser, K. Synuclein Family Members Prevent Membrane Damage by Counteracting α-Synuclein Aggregation. Biomolecules 2021, 11, 1067. https://doi.org/10.3390/biom11081067
Scheibe C, Karreman C, Schildknecht S, Leist M, Hauser K. Synuclein Family Members Prevent Membrane Damage by Counteracting α-Synuclein Aggregation. Biomolecules. 2021; 11(8):1067. https://doi.org/10.3390/biom11081067
Chicago/Turabian StyleScheibe, Christian, Christiaan Karreman, Stefan Schildknecht, Marcel Leist, and Karin Hauser. 2021. "Synuclein Family Members Prevent Membrane Damage by Counteracting α-Synuclein Aggregation" Biomolecules 11, no. 8: 1067. https://doi.org/10.3390/biom11081067
APA StyleScheibe, C., Karreman, C., Schildknecht, S., Leist, M., & Hauser, K. (2021). Synuclein Family Members Prevent Membrane Damage by Counteracting α-Synuclein Aggregation. Biomolecules, 11(8), 1067. https://doi.org/10.3390/biom11081067