1. Introduction
Gallium-68 (
68Ga) is a positron-emitting radioisotope with a half-life of 68 min. This relatively short half-life is suitable for positron emission tomography (PET) diagnostics when radiolabeling small molecules with fast pharmacokinetics [
1,
2].
The 68Ge/68Ga-generators are easy to use but do suffer from several drawbacks. The small amount of radioactivity that can be eluted (e.g., ~1.5 GBq from a new generator) requires multiple generators to scale up radiopharmaceutical production. After elution, several hours are needed for 68Ga ingrowth before satisfactory radioactivity levels can be eluted again. This means the generator may be used for radiolabeling 2–3 times a day during normal working hours. Additionally, decay of the parent 68Ge leads to elution of less and less 68Ga radioactivity over the generator lifespan. If 3–4 patient doses per batch are typically obtained when the generator is new, only 1–2 doses per batch are produced toward the end of its use. The worldwide demand for 68Ga-based radiopharmaceuticals is constantly increasing, particularly since the successful introduction of theranostics, in which 68Ga is the diagnostic radionuclide. Although the commercial production of generators has recently expanded, there is still an unmet need for 68Ga, with long delivery times and relatively high pricing. Another drawback is the need to store expired generators for several years before destruction due to the 271-day half-life of germanium-68 (68Ge).
The approval of
68Ge/
68Ga-generators has tremendously facilitated the clinical implementation of several
68Ga-labelled tracers in the last five years. The availability of
68Ge/
68Ga-generators has had undoubted importance for the development of new radiopharmaceuticals for preclinical applications to clinical implementation, as well as for enabling hospitals without access to a cyclotron to produce their own PET radiopharmaceuticals [
2,
3].
Altogether, these limiting factors have driven the development of alternatives to
68Ge/
68Ga-generators to improve the availability of
68Ga [
4]. The ability to produce
68Ga with a low-energy cyclotron is an important development and has recently been clinically implemented [
5].
The cyclotron production of
68Ga is feasible using either liquid or solid targets. In liquid-target productions, a solution of enriched
68Zn salt is irradiated to produce the desired radiometal via the
68Zn(p,n)
68Ga reaction [
6,
7]. The zinc solution to be irradiated is conveniently transferred through transfer lines to the target holder before irradiation and also after irradiation to the synthesis hot cell for purification and radiolabeling in the radiopharmaceutical production. The yields of radiopharmaceuticals produced from liquid target
68Ga are similar or slightly increased compared to those using a
68Ge/
68Ga-generator.
Production of
68Ga by the
68Zn(p,n)
68Ga reaction using solid-target systems on low-energy medical cyclotrons [
8,
9,
10] has yielded the highest radioactivity, up to 370 GBq [
11]. In both liquid- and solid-target productions, it is of critical importance to separate
68Ga from the irradiated
68Zn and other metal ions. Many separation techniques have been suggested [
12]. Incompletely removed metal ion impurities compete with Ga
3+ in the complexation with different chelators, which negatively affects the radiolabeling yields. In addition to the zinc that must be removed, predominantly, the metal ion of concern is the trivalent Fe
3+ [
13], which has a higher stability constant (log K
ML) for the chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-acetic acid (DOTA), for example, than Ga
3+ [
14,
15].
Our group has recently developed a solid-target
68Ga production purification sequence based on double anion exchange Uranium and TEtraValents Actinides (UTEVA
®) resin columns, washed in an effective last step with hydrochloric acid (HCl) (2.5 N) to minimize the content of Zn
2+ (target material) ions remaining in the
68GaCl
3 eluate [
13]. All quality requirements, according to the European Pharmacopoeia monograph for cyclotron produced
68Ga [
16], were fulfilled. An apparent molar activity (AMA) of 86 ± 22 GBq/µmol (
n = 3), determined by DOTA titrations, was achieved. The content of Zn in the eluate (Zn to activity ratio) was satisfying, setting the shelf-life of the
68GaCl
3 eluate to 7.7 h. The limiting factor was the content of Fe in the eluate (Fe to activity ratio), which set the final shelf-life of the
68GaCl
3 eluate to 6.4 h. However, when using this
68GaCl
3 eluate for radiopharmaceutical productions of [
68Ga]Ga-DOTATOC or [
68Ga]Ga-FAPI-46, an RCY of only approximately 25% were obtained, with a 40 or 50 µg precursor, respectively. From 10 GBq
68GaCl
3 eluate, 2.5 GBq product was obtained. Although this means a three-fold higher product activity compared to generator-produced syntheses, there is still a large fraction of radioactivity that is lost during the synthesis. This prompted us to make further improvements to increase the RCY.
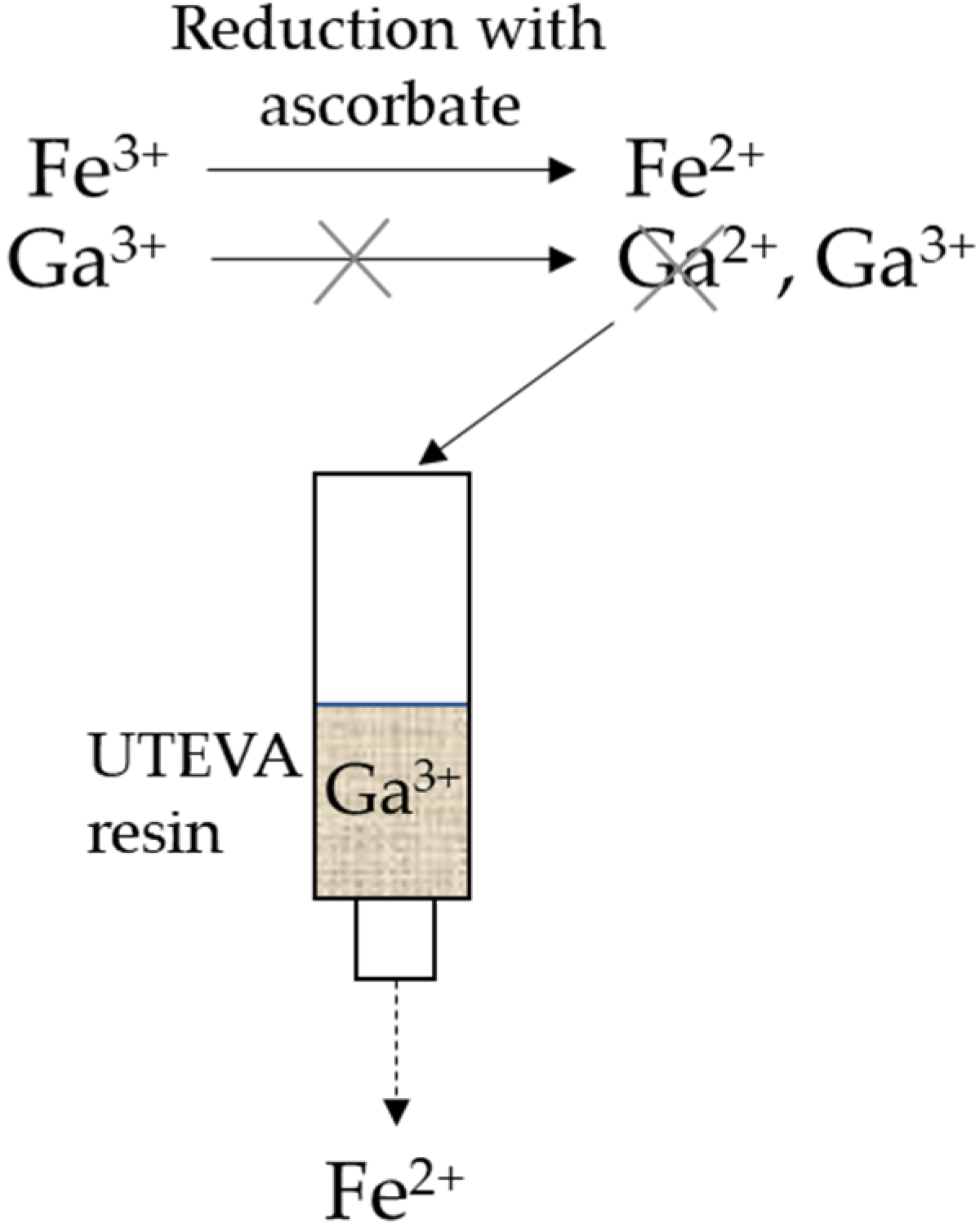
We hypothesized that decreasing the amount of Fe3+ in the cyclotron-produced 68GaCl3 eluate would increase the RCY, and consequently, the AMA.
It is important to take into account that the metal ions are in constant equilibrium with the surrounding negatively charged counter ions and water molecules that act as ligands and form metal complexes. The speciation of the metal complexes is of crucial importance due to the charge and electrostatic interactions with the surrounding environment. High concentrations of chloride, and low pH, favor the formation of negatively charged complexes, such as [FeCl
4]
− and [GaCl
4]
−. The negatively charged complexes follow the HCl concentration and the distribution coefficients of the metal ions for the UTEVA resin [
17,
18,
19]. The active part of the UTEVA resin consists of a neutral dipentyl pentylphosphonate complexing ligand for the metal ion [
17]. The reduction potential of Fe
3+ to Fe
2+ at a low pH is around +0.8 V and of Ga
3+ to Ga
2+ at around −0.6 V [
20,
21]. The oxidation potential of ascorbic acid at low pH is around −0.3 V [
22]. Due to the lower reduction potential of Ga
3+, which is lower than the oxidation potential of ascorbic acid, the result is a reduction of Fe
3+ to Fe
2+, while gallium is kept in the form of Ga
3+. See
Figure 1 below.
The aim of this work was to improve the purification methodology of the solid target production of 68Ga. By adding ascorbate to the purification steps, the level of Fe3+ in the 68GaCl3 eluate was significantly decreased; thus, enabling high-yield radiolabeling of clinically relevant DOTA-based tracers, such as DOTATOC and FAPI-46.
2. Materials and Methods
2.1. 68Ga Production and Purification Modification with Added Ascorbate
68GaCl
3 was produced via the
68Zn(p,n)
68Ga and purified according to our previous method [
13], except with an addition of 500 mg of sodium ascorbate (Apotekets Produktion och Laboratorier (APL), Stockholm, Sweden), divided between the HCl dilution and wash solutions as shown in
Figure 2. In short, 110 mg
68Zn enriched (98.7 ± 0.2%) foil (Isoflex, San Francisco, CA, USA) was pneumatically transferred using a transfer module (Comecer EDS) to the cyclotron’s irradiation station (GE Healthcare, Uppsala, Sweden, PETtrace 800 and Comecer PTS). Irradiation was performed with a proton beam current of 25 µA for 68 min. Dissolution and separation were fully automated using a cassette-based Taddeo PRF module (Comecer, Castel Bolognese, Italy), and all materials and acids used were of metal-free quality, as stated in [
13]. The RNP of the eluate was determined by gamma spectroscopy using a high-purity germanium detector (Canberra with Cryo-Cycle II Hybrid Cryostat), radionuclidic identity was determined by half-life measurement using a dose calibrator (Capintec CRC-55tR, LabLogic, Sheffield, United Kingdom), as described in [
13].
2.2. Colorimetric Test of Iron Content and ICP-MS Measurements
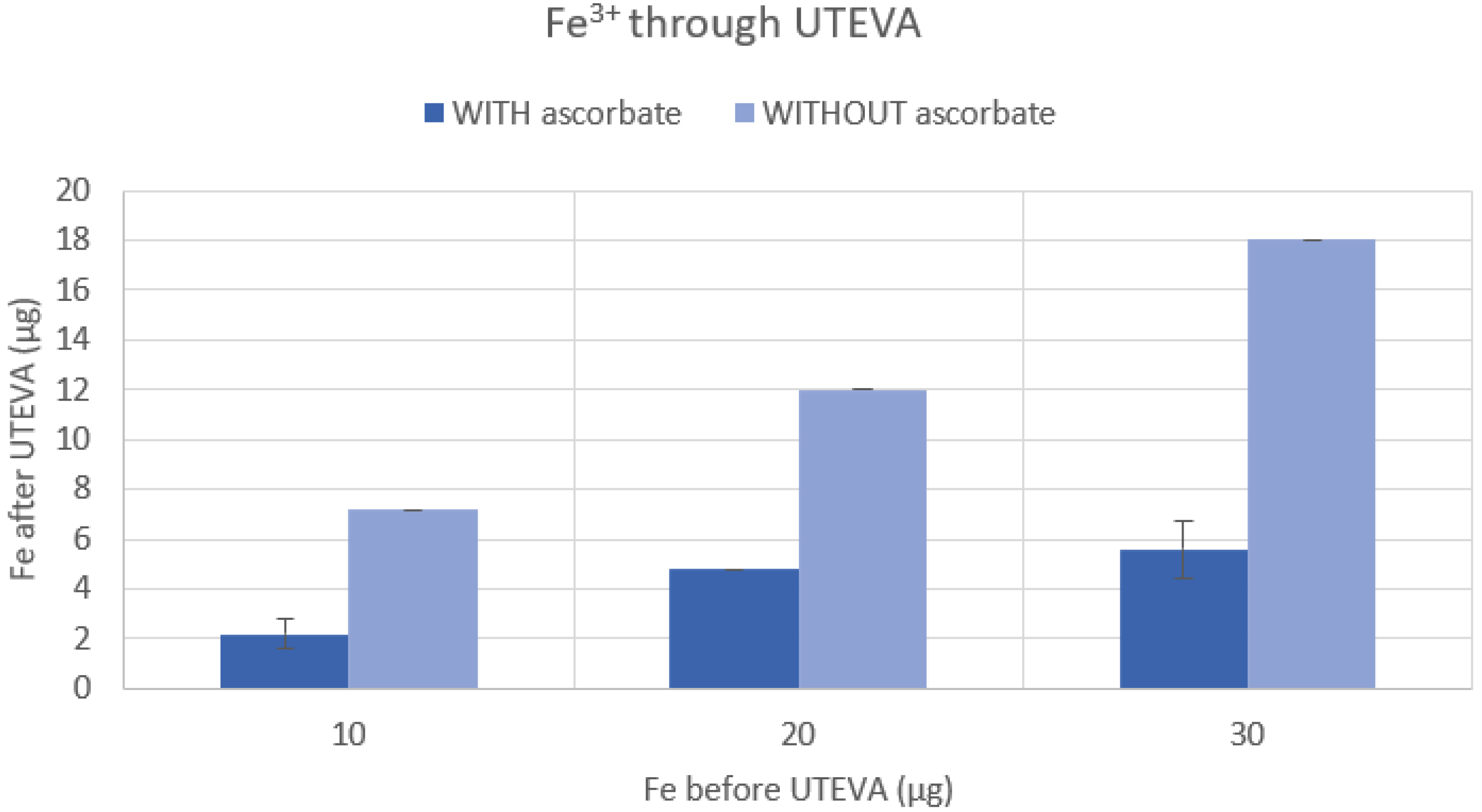
To investigate that iron was eliminated to a larger extent when reduced to Fe2+ using ascorbate in the UTEVA resin purification method, we first performed a cold colorimetric measurement of the content of iron present in the rinses with or without the addition of ascorbate. The UTEVA resin (110 mg) was loaded with Fe3+ (Fe(III)Cl3, Sigma-Aldrich, Stockholm, Sweden), rinsed, and eluted in a fashion comparable to that used in the 68Ga purification of this study. The UTEVA resin was conditioned with HCl (4 N, 4 mL), 10/20/30 µg Fe3+ in HCl (4 N, 2 mL) (with or without 10 mg/mL ascorbate) was loaded and trapped on the resin, following rinses with HCl (4 N, 10 mL) (with or without 10 mg/mL ascorbate) and HCl (2.5 N, 8 mL) (with or without 10 mg/mL ascorbate), and lastly dried with 20 mL of air. The resin was then eluted using 1 mL of water (TraceSelect, Honeywell, Seetze, Germany), and the eluate was collected for analysis of iron content using an iron colorimetric test (MColortest, part no. 1.14759.0001, Merck, Darmstadt, Germany). The p-values were calculated using the Student’s t-test in Excel (Microsoft® Excel® for Microsoft 365MSO); p < 0.05 was considered statistically significant.
68GaCl3 eluate from one 68Ga solid-target cyclotron production with added ascorbate, and one without ascorbate, were analyzed by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) externally (ALS, Umeå, Sweden). The analysis included the following metal ions, Zn (calibrated for 68Zn instead of natZn), Fe, Ga, Al, Cd, Cu, Ge, Mo, Ni, Pb, Pt, and Ti.
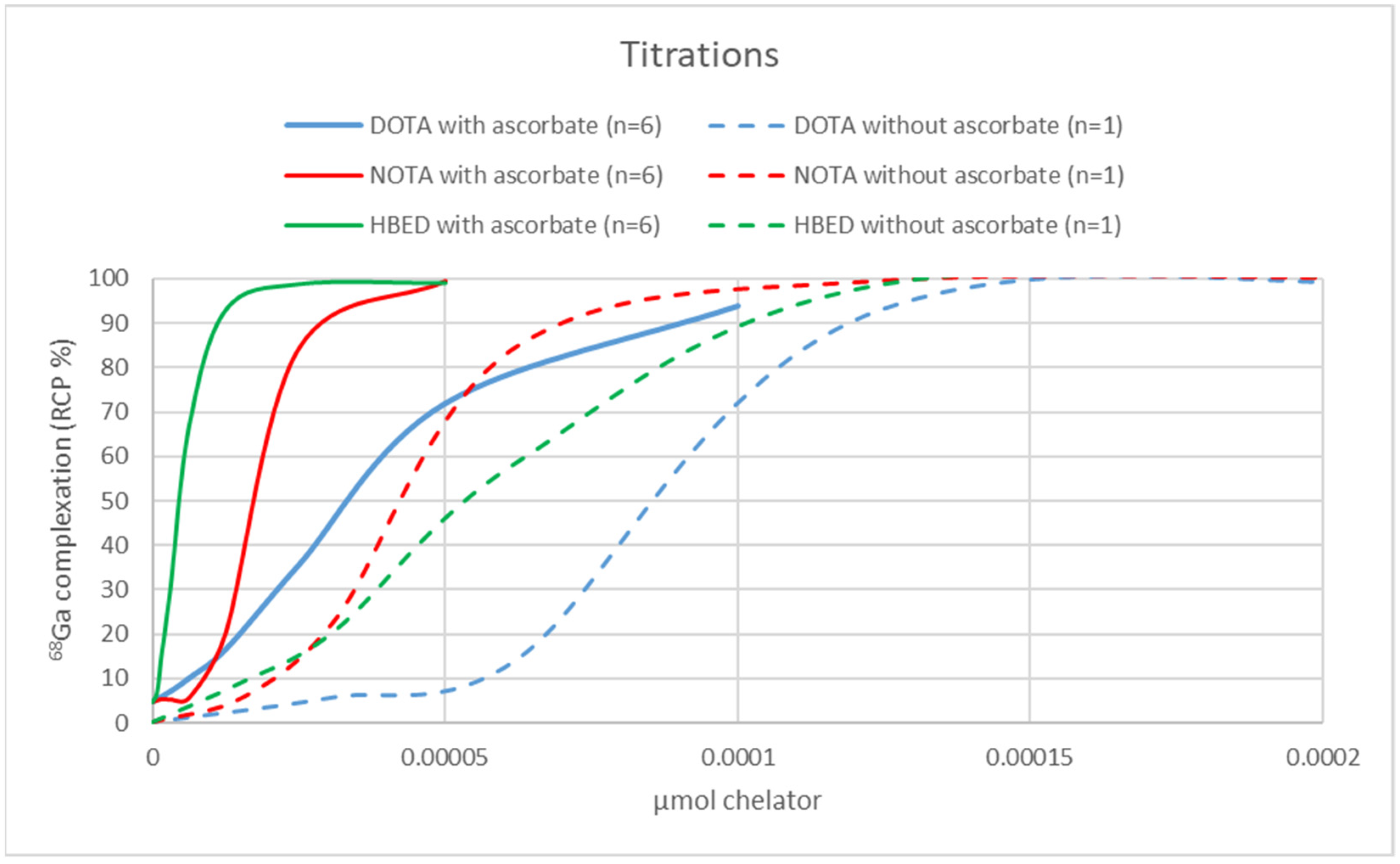
2.3. Chelator Titrations and AMA Determination
AMA of the cyclotron-produced
68GaCl
3 eluate was determined on 50 µL (5% of total eluate volume) by titration with the chelators DOTA (Sigma-Aldrich), (1,4,7-triazonane-1,4,7-triyl) triacetic acid (NOTA) (CheMatech, Dijon, France), and
N,N′-Di(2-hydroxybenzyl)ethylenediamine-
N,N′-diacetic acid monohydrochloride hydrate (HBED) (STEM Chemicals Inc., Bischheim, France). Chelator solutions were prepared in serial dilutions. Ranges of chelators labeled in the titrations, when ascorbate was used in the purification: DOTA 3.1 pmol–0.1 nmol, NOTA and HBED 1.2 pmol–0.05 nmol, and when ascorbate was not used in the purification: DOTA 31.2 pmol–1.0 nmol, NOTA and HBED 15.5 pmol–0.5 nmol. The
68GaCl
3 solution was adjusted to pH 4.0 using sodium acetate buffer (~1:10 acetate buffer solution pH 4.6 (Honeywell Fluka, Steinheim, Germany) in TraceSelect Water (Honeywell) pH adjusted with HCl (Honeywell)) to a final volume of 600 µL in each vial. The vials were incubated at 95 °C, 550 rpm for 15 min (Eppendorf (ThermoMixer C)). The AMA was analyzed by measuring the labeling efficiency of each vial. Analysis of labeling efficiency (incorporation of
68Ga in DOTA, NOTA, and HBED) was performed by radio-thin layer chromatography using iTLC-SG-strip (Agilent, Folsom, CA, USA) as stationary phase, eluted in ammonium acetate 1 M (Sigma-Aldrich): methanol (Merck) 1:1 as mobile phase. In this analysis, free
68Ga stayed at the origin (Retardation factor, Rf~0–0.1) while complexed
68Ga-DOTA,
68Ga-NOTA, and
68Ga-HBED migrated (Rf~0.9–1.0). Radioactivity in the strips was detected by a TLC-scanner (AR-2000, Eckert & Ziegler, Berlin, Germany), and analysis was performed using the software WinScan 3.0 (Eckert & Ziegler). Labeling efficiency was plotted as a function of DOTA, NOTA, and HBED chelator mass (µmol). AMA was calculated by the equation of the line and determined as 50% incorporation and by dividing these values by two, as suggested earlier [
23]. The values were decay-corrected to the end of
68GaCl
3 eluate purification (EOP).
2.4. Synthesis of [68Ga]Ga-FAPI-46 and [68Ga]Ga-DOTATOC
From each cyclotron production of 68GaCl3 (total volume of ~1 mL), 50 µL of the eluate was used for chelator titration as described above. The remainder of the radioactivity (~11 GBq) was used for each radiopharmaceutical synthesis.
Automated radiosynthesis was performed on an Eckert & Ziegler Modular-Lab PharmTracer synthesis module using the Modular-Lab software (Eckert & Ziegler). See
Figure 3 for a schematic flow diagram of the synthesis. All materials used for radiolabeling were of GMP grade and metal-free quality if not otherwise stated. All buffer kits and hardware kits (synthesis cassettes) for the syntheses were purchased from Eckert & Ziegler.
The
68GaCl
3 eluate was diluted to 4–5 mL with 0.1 N HCl (Eckert & Ziegler) to minimize activity losses in the synthesis cassette-connected eluate transfer tube. The reaction vessel was prepared to contain 50 µg of FAPI-46 precursor (Sofie Biosciences, Totowa, NJ, USA) or 40 µg of DOTATOC precursor (ABX, advanced biochemical compounds, Radeberg, Germany) and buffer solution (54 mg sodium acetate trihydrate, 18 µL 30% HCl, 8 µL glacial acetic acid, 2.4 mL TraceSelect water, and 0.2 mL ethanol). The diluted
68GaCl
3 eluate was transferred to the synthesis unit and trapped on a cationic exchange cartridge (SCX in the synthesis scheme,
Figure 3a) and eluted into the reaction vial with 0.7 mL of sodium chloride (NaCl) 5 N/HCl 0.13 N. The final volume of the reaction mixture was 3.3 mL, pH 3.5. The labeling reaction mixture was heated to 95 °C for 5 min. After the end of the labeling, the crude product was diluted with 2 mL of 4 mg/mL sodium ascorbate in 9 mg/mL NaCl and trapped on a reversed-phase solid-phase extraction (SPE) cartridge (C18 in the synthesis scheme,
Figure 3a). The SPE was rinsed to waste using 4 mL of 4 mg/mL sodium ascorbate in 9 mg/mL NaCl to remove any remaining free
68Ga ions in the system. The trapped product was then eluted from the SPE, using 1.2 mL of ethanol/water 1:1, through a 0.22 µm sterile filter (Millex-GV, Merck Millipore, Darmstadt, Germany) into the product vial. The product ([
68Ga]Ga-FAPI-46 or [
68Ga]Ga-DOTATOC) was lastly diluted with 4 mg/mL sodium ascorbate (APL, Sweden), as a radiolytic stabilizer, in 9 mg/mL NaCl to a final formulation volume of approximately 9.5 mL. From the addition of eluate to the finished product, the time required was 17 min. Radiosynthesis of these products using generator-based
68GaCl
3 eluate was performed in the same way, using eluate from a GalliaPharm generator (Eckert & Ziegler) or a GalliAd generator (IRE ELiT, Fleurus, Belgium). The synthesis of [
68Ga]Ga-DOTATOC was, however, performed without ascorbate as a stabilizer. A flow diagram of the syntheses is illustrated in
Figure 3b.
2.5. Quality Control of [68Ga]Ga-FAPI-46 and [68Ga]Ga-DOTATOC
Full quality controls (QC) were performed for [68Ga]Ga-FAPI-46 and [68Ga]Ga-DOTATOC using qualified instruments if not otherwise stated. The QC attributes determined included the appearance by visual inspection and pH by pH strip (Merck, Darmstadt, Germany). The content of bacterial endotoxins was performed by chromogenic LAL-test method using Endosafe-Nextgen PTS (Charles River, Willmington, MA, USA), and the filter integrity was tested by a bubble point tester (DM Automation, Sweden or an in-house built, qualified bubble point tester).
The radiochemical purity (RCP), chemical purity, as well as radiochemical stability were measured by analytical radio-high performance liquid chromatography (radio-HPLC). Two different HPLC systems were used. The Agilent 1260 Infinity System is equipped with a quaternary pump, autosampler, and DAD UV detector (254 nm) as well as a FlowRAM 2”NaI/PMT radiodetector (LabLogic, Sheffield, United Kingdom) and the software Laura (LabLogic) was used for [68Ga]Ga-FAPI-46. Analysis was performed on an analytical column (Agilent Poroshell 120 EC-C18, 2.7 µm 4.6 × 100 mm) and a guard column (Poroshell 120 EC-C18 Fast guard, 3 × 5 mm, 2.7 µm). The mobile phase was a gradient composed of 50 mM phosphoric acid (H3PO4) and acetonitrile (CH3CN); a flow rate of 0.3 mL/min was used.
The Shimadzu HPLC system (Duisburg, Germany) is equipped with a binary pump, degasser (Biotech, Onsala, Sweden), manual injector (Rheodyne, Bensheim, Germany), and UV-VIS detector (220 nm), as well as a radiodetector (Bioscan, Washington, DC, USA) and the software Shimadzu LC Solution was used for [68Ga]Ga-DOTATOC. Analysis was performed using an analytical column (ACE 3-C18, 4.6 × 150 mm) and a guard column of the same material (3 µm). The mobile phase was a gradient composed of 0.1% TFA in CH3CN:H2O, and a flow rate of 0.6 mL/min was used.
The radiochemical impurities of 68Ga ions and 68Ga-colloids were determined with iTLC analysis using iTLC-SG strip (Agilent). The radioactivity was detected using a radio-TLC scanner, either Scan-RAM with a PS/PMT detector, equipped with the software Laura (LabLogic) or the Bioscan TLC scanner, equipped with the software Winscan (Bioscan). The mobile phase of 5 M ammonium acetate (Merck) and methanol (Merck) in a ratio of 25:75 was used for [68Ga]Ga-FAPI-46 while the mobile phase of 1 M ammonium acetate (Merck) and methanol (Merck) in a ratio 1:1 was used for [68Ga]Ga-DOTATOC. In these systems, Rf was ~0–0.2 for 68Ga-impurities, and Rf was ~0.6–1.0 for 68Ga-labeled products.
Ethanol levels in the products were analyzed using a gas chromatograph (GC model 6850 Agilent) equipped with a flame ionization detector, an Agilent Res-Solv column (30 m × 0.53 mm ID × 1.0 µm film), and an autoinjector. The GC method used a 2 µL injection volume, a split ratio of 1:80, and helium as a carrier gas. The temperature was programmed to 35 °C for 3.5 min after injection, ramped to 240 °C at a rate of 70 °C/min, held at 240 °C for 3 min, and cooled to 35 °C.
The stability (shelf-life) of 68Ga-labeled products was determined by analyzing the total radiochemical purity of the product with HPLC and iTLC as described above. Sterility tests were performed by direct inoculation by an external contractor (APL, Stockholm, Sweden).
4. Discussion
The feasibility to utilize all high out-put cyclotron-produced 68GaCl3 eluate for radiolabeling of DOTA-based tracers is highly dependent on the purity (i.e., content of competing metal ions impurities) of the 68Ga-eluate. Impurities may originate from the starting materials, i.e., dilution and wash solutions, tubing, and especially the target material (i.e., the 68Zn foil).
Low content of competing ions such as zinc and iron, especially the trivalent Fe
3+, when for example, DOTA is used as a chelator in the radiolabeling, is of great importance. The importance of decreasing the content of Fe
3+ in the
68GaCl
3 eluate is the metal ions’ higher stability constant in association with the chelator, i.e., its’ ability to form stable complexes with the chelator [
14,
15].
In this study, we have demonstrated a straight-forward and improved purification approach to achieve a cyclotron solid target produced
68GaCl
3 eluate with significantly lower levels of competing metal ions (i.e., Fe
3+) by the addition of ascorbate. Ascorbate is a powerful antioxidant that is commonly used as a radiolytic stabilizer in radiopharmaceuticals [
24,
25]. Here, by utilizing sodium ascorbate’s predominant ability to reduce Fe
3+ to Fe
2+ and its inability to reduce Ga
3+ to Ga
2+, a more effective separation through the UTEVA resins is possible in the purification process. This enables the use of cyclotron-produced
68GaCl
3 eluate for high-yield DOTA-chelate complexation with superior results.
AMA values, analyzed by titrations, were 2-, 3- and 16-fold higher for DOTA, NOTA, and HBED, respectively, verifying a considerable improvement. The difference in the AMA increase for the different chelators might possibly be explained by their binding stabilities and stability constants (log K
ML) to Ga
3+, Fe
3+ and Fe
2+, as further summarized in
Table S2, in the Supplementary section. The incredible increase in AMA for HBED may be related to its high log K
ML to both Ga
3+ and Fe
3+. Notably, this purification approach has enabled high-yield DOTA-based radiopharmaceutical productions of 5.58 ± 0.35 GBq (
n = 3) for [
68Ga]Ga-FAPI-46 and 6.1 ± 1.3 GBq (
n = 3) for [
68Ga]Ga-DOTATOC.
High concentrations of competing metal ions may be, alternatively, compensated for by increasing the amounts of precursor used in the radiolabeling, as previously demonstrated [
9,
11]. For example, Tieu et al. [
9] used 80 µg of DOTATATE precursor for labeling with 6.3 GBq
68GaCl
3 and received 3.31 GBq [
68Ga]Ga-DOTA-TATE (RCY = 70%, RCP = 68%). Thisgaard et al. [
11] used 500 µg and received 3.22 GBq [
68Ga]Ga-DOTA-TATE product. This approach will increase the produced radioactivity but will also lower the AMA. This could be problematic due to the restricted maximum peptide dose allowable for patient administration according to the European Pharmacopoeia (e.g., 50 µg of DOTATOC [
26]). This would consequently limit the shelf life of the radiopharmaceutical product.
Currently, there is no clinical establishment defining the influence of AMA on the imaging utility of
68Ga-labeled tracers in oncological applications. It is, however, known from generator-produced batches that clinical imaging is feasible in the AMA ranges of 7–25 GBq/µmol. The impact of AMA has been closely investigated in some limited preclinical studies. Lin et al. reported that in vitro cell uptake and better contrast in in vivo preclinical imaging was seen with increasing AMA of [
68Ga]Ga-PSMA-11 [
27]. Increased in vitro cell uptake with AMA was also reported for the same radiotracer by Sanchez-Crespo et al. [
28]. In a study by von Hacht et al., the low AMA of the DOTA-based
68Ga-labeling was resolved by preparative HPLC purification, thereby improving the detection of small metastases [
29]. The level of AMA and its impact in diagnostics is an interesting and important aspect, which is made available also for DOTA-based
68Ga-labeled radiopharmaceuticals by the results from this present study.
It is of considerable interest to be able to utilize cyclotron-produced 68Ga eluate in kit preparations of 68Ga-based tracers, as more kits are elegantly prepared for one single vial compounding in which the eluate is directly added, thereby minimizing radiation exposure and handling. The high AMA cyclotron-produced 68Ga eluate obtained here warrants/can facilitate future kit preparation procedures.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}