
BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme


Abstract
:1. Introduction
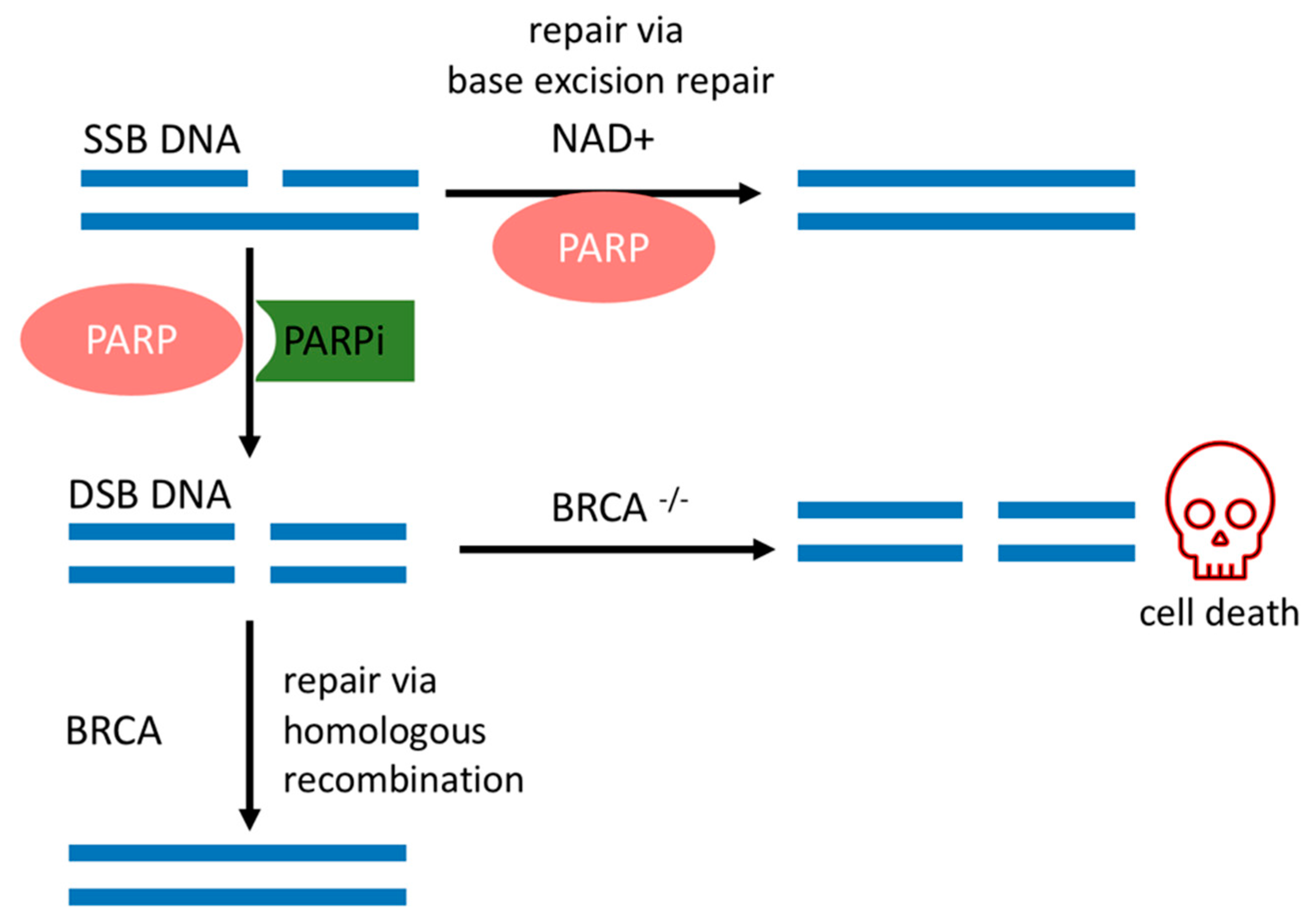
2. PARPi and Synthetic Lethality
3. GBM: Brief Overview
4. BRCAness Biomarkers in GBM
4.1. IDH-1/2
4.2. EGFR vIII
4.3. PTEN
4.4. MYC
4.5. ERβ
5. Clinical Trials and Biomarkers
6. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Chow, D.; Chang, P.; Weinberg, B.D.; Bota, D.A.; Grinband, J.; Filippi, C.G. Imaging Genetic Heterogeneity in Glioblastoma and Other Glial Tumors: Review of Current Methods and Future Directions. Am. J. Roentgenol. 2017, 210, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Friedmann-Morvinski, D. Glioblastoma Heterogeneity and Cancer Cell Plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 2019, 8, 670. [Google Scholar] [CrossRef] [Green Version]
- Boukerroucha, M.; Josse, C.; Segers, K.; El-Guendi, S.; Frères, P.; Jerusalem, G.; Bours, V. BRCA1 germline mutation and glioblastoma development: Report of cases. BMC Cancer 2015, 15, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terkelsen, T.; Christensen, L.-L.; Fenton, D.C.; Jensen, U.B.; Sunde, L.; Thomassen, M.; Skytte, A.-B. Population frequencies of pathogenic alleles of BRCA1 and BRCA2: Analysis of 173 Danish breast cancer pedigrees using the BOADICEA model. Fam. Cancer 2019, 18, 381–388. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Ning, J.-F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Bürkle, A. Poly(APD-ribosyl)ation, a DNA damage-driven protein modification and regulator of genomic instability. Cancer Lett. 2001, 163, 1–5. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Shar-yin, N.H.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Park, J.K.; Hodges, T.; Arko, L.; Shen, M.; Dello Iacono, D.; McNabb, A.; Olsen Bailey, N.; Kreisl, T.N.; Iwamoto, F.M.; Sul, J.; et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J. Clin. Oncol. 2010, 28, 3838–3843. [Google Scholar] [CrossRef]
- Faith, G.D.; Sally, F.; James, G.; Suna, B.; Steven, B. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: An analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973–1991. J. Neurosurg. 1998, 88, 1–10. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Lewis, A.D.; Fitzsimmons, S.A.; Workman, P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int. J. Radiat. Oncol. 1994, 29, 427–431. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Jihong, Z.; Malcolm, F.G.S.; Tracey, D.B. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Leonetti, C.; Biroccio, A.; Graziani, G.; Tentori, L. Targeted Therapy for Brain Tumours: Role of PARP Inhibitors. Curr. Cancer Drug Targets 2012, 12, 218–236. [Google Scholar] [CrossRef] [PubMed]
- Ron, B.; Asna, N.; Pamela, S.; Nicole, F.; Moshe, S. Glioblastoma Multiforme, Diagnosis and Treatment; Recent Literature Review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration, FDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125085s0169lbl.pdf (accessed on 16 July 2021).
- Kaka, N.; Hafazalla, K.; Samawi, H.; Simpkin, A.; Perry, J.; Sahgal, A.; Das, S. Progression-Free but No Overall Survival Benefit for Adult Patients with Bevacizumab Therapy for the Treatment of Newly Diagnosed Glioblastoma: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 1723. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Murnyák, B.; Kouhsari, M.C.; Hershkovitch, R.; Kálmán, B.; Marko-Varga, G.; Klekner, Á.; Hortobágyi, T. PARP1 expression and its correlation with survival is tumour molecular subtype dependent in glioblastoma. Oncotarget 2017, 8, 46348–46362. [Google Scholar] [CrossRef] [Green Version]
- Bandey, I.; Chiou, S.H.; Huang, A.P.; Tsai, J.C.; Tu, P.H. Progranulin promotes Temozolomide resistance of glioblastoma by orchestrating DNA repair and tumor stemness. Oncogene 2015, 34, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clin. Cancer Res. 2007, 13, 2728. [Google Scholar] [CrossRef] [Green Version]
- Muscal, J.A.; Thompson, P.A.; Giranda, V.L.; Dayton, B.D.; Bauch, J.; Horton, T.; McGuffey, L.; Nuchtern, J.G.; Dauser, R.C.; Gibson, B.W.; et al. Plasma and cerebrospinal fluid pharmacokinetics of ABT-888 after oral administration in non-human primates. Cancer Chemother. Pharmacol. 2010, 65, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Mikule, K.; Wang, Z.; Poon, G.; Vaidyanathan, A.; Smith, G.; Zhang, Z.-Y.; Hanke, J.; Ramaswamy, S.; Wang, J. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models. Oncotarget 2018, 9, 37080–37096. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: Results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1306–1315. [Google Scholar] [CrossRef]
- Louis, D.N.; Arie, P.; Guido, R.; von Deimling, A.; Dominique, F.-B.; Webster, K.C.; David, W.E. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Hakin-Smith, V.; Jellinek, D.A.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.R.; McKay, M.J.; Reddel, R.R.; Royds, J.A. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef]
- Mukherjee, J.; Johannessen, T.-C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, J.; Dalle-Ore, C.; Johanessen, T.-C.; Pandita, A.; Ohba, S.; Cahill, D.; Pieper, R.O. Cbmt-19. The alternative lengthening of telomere (alt) mechanism provides collateral sensitivity to lethal telomeric fusion induced by trapping parp inhibitors. Neuro Oncol. 2019, 21, vi37. [Google Scholar] [CrossRef]
- Garbarino, J.; Eckroate, J.; Sundaram, R.K.; Jensen, R.B.; Bindra, R.S. Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl. Oncol. 2021, 14, 101147. [Google Scholar] [CrossRef]
- George, S.L.; Lorenzi, F.; King, D.; Hartlieb, S.; Campbell, J.; Pemberton, H.; Toprak, U.H.; Barker, K.; Tall, J.; da Costa, B.M.; et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 2020, 59, 102971. [Google Scholar] [CrossRef]
- Ducray, F.; Marc, S.; Olivier, L.; Chinot, M.F.; Romain, R.; Laure, T.-M.; POLA Network. Olaparib in Recurrent Idh-Mutant High-Grade Glioma (Olagli). J. Clin. Oncol. 2021, 15, 2007. [Google Scholar] [CrossRef]
- McLendon, R.E.; Turner, K.; Perkinson, K.; Rich, J. Second messenger systems in human gliomas. Arch. Pathol. Lab. Med. 2007, 131, 1585–1590. [Google Scholar] [CrossRef]
- Li, B.; Yuan, M.; Kim, I.-A.; Chang, C.-M.; Bernhard, E.J.; Shu, H.-K.G. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 2004, 23, 4594–4602. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lee, J.H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, B.; McEllin, B.; Camacho, C.V.; Tomimatsu, N.; Sirasanagandala, S.; Nannepaga, S.; Hatanpaa, K.J.; Mickey, B.; Madden, C.; Maher, E.; et al. EGFRvIII and DNA Double-Strand Break Repair: A Molecular Mechanism for Radioresistance in Glioblastoma. Cancer Res. 2009, 69, 4252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Gao, F.; Zheng, S.; Zhang, C.; Martinez-Ledesma, E.; Ezhilarasan, R.; Ding, J.; Li, X.; Feng, N.; Multani, A.; et al. EGFR amplification induces increased DNA damage response and renders selective sensitivity to Talazoparib (PARP inhibitor) in glioblastoma. Clin. Cancer Res. 2019, 26, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.-S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Majuelos-Melguizo, J.; Rodríguez, M.I.; López-Jiménez, L.; Rodríguez-Vargas, J.M.; Martí Martín-Consuegra, J.M.; Serrano-Sáenz, S.; Gavard, J.; de Almodóvar, J.M.R.; Javier Oliver, F. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget 2014, 6, 4790–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; de Gooijer, M.C.; Roig, E.M.; Buil, L.C.M.; Christner, S.M.; Beumer, J.H.; Würdinger, T.; Beijnen, J.H.; van Tellingen, O. ABCB1, ABCG2, and PTEN Determine the Response of Glioblastoma to Temozolomide and ABT-888 Therapy. Clin. Cancer Res. 2014, 20, 2703. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, A.J.; Jackson, A.; Swaisland, H.; Stewart, W.; Halford, S.E.R.; Molife, L.R.; Hargrave, D.R.; McCormick, A. Results of stage 1 of the oparatic trial: A phase I study of olaparib in combination with temozolomide in patients with relapsed glioblastoma. J. Clin. Oncol. 2014, 32, 2025. [Google Scholar] [CrossRef]
- Sewastianik, T.; Prochorec-Sobieszek, M.; Chapuy, B.; Juszczyński, P. MYC deregulation in lymphoid tumors: Molecular mechanisms, clinical consequences and therapeutic implications. Biochim. Biophys. Acta (BBA) Bioenerg. Rev. Cancer 2014, 1846, 457–467. [Google Scholar] [CrossRef]
- Hui, A.B.-Y.; Lo, K.-W.; Yin, X.-L.; Poon, W.-S.; Ng, H.-K. Detection of Multiple Gene Amplifications in Glioblastoma Multiforme Using Array-Based Comparative Genomic Hybridization. Lab. Investig. 2001, 81, 717–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, S.; Koehler, K.F.; Gustafsson, J.-Å. Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 2011, 10, 778–792. [Google Scholar] [CrossRef]
- Liu, J.; Sareddy, G.R.; Zhou, M.; Viswanadhapalli, S.; Li, X.; Lai, Z.; Tekmal, R.R.; Brenner, A.J.; Vadlamudi, R.K. Differential effects of estrogen receptor beta isoforms on glioblastoma progression. Cancer Res. 2018, 78, 3176–3189. [Google Scholar] [CrossRef] [Green Version]
- Sareddy, G.R.; Nair, B.C.; Gonugunta, V.K.; Zhang, Q.G.; Brenner, A.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. Therapeutic significance of estrogen receptor β agonists in gliomas. Mol. Cancer Ther. 2012, 11, 1174–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiee, M.; Mafi, A.; Nilipour, Y.; Sourati, A.; Sassanpour, P.; Tabatabaeefar, M. Estrogen Receptor Expression in Glial Tumors of Iranian Patients: A Single Center Experience. Iran J. Pathol. 2020, 15, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Sareddy, G.R.; Li, M.; Liu, J.; Luo, Y.; Venkata, P.P.; Viswanadhapalli, S.; Tekmal, R.R.; Brenner, A.; Vadlamudi, R.K. Estrogen receptor beta enhances chemotherapy response of GBM cells by down regulating DNA damage response pathways. Sci. Rep. 2019, 9, 6124. [Google Scholar] [CrossRef]
- Jiang, X.; Li, W.; Li, X.; Bai, H.; Zhang, Z. Current status and future prospects of PARP inhibitor clinical trials in ovarian cancer. Cancer Manag. Res. 2019, 11, 4371–4390. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.E.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.M.; Armstrong, A.; et al. OlympiAD: Phase III trial of olaparib monotherapy versus chemotherapy for patients (pts) with HER2-negative metastatic breast cancer (mBC) and a germline BRCA mutation (gBRCAm). J. Clin. Oncol. 2017, 35, LBA4. [Google Scholar] [CrossRef]
- Food and Drug Administration, FDA. Fda Approves Olaparib for Hrr Gene-Mutated Metastatic Castration-Resistant Prostate Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer (accessed on 16 July 2021).
- Food and Drug Administration, FDA. Fda Approves Olaparib for Gbrcam Metastatic Pancreatic Adenocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-gbrcam-metastatic-pancreatic-adenocarcinoma (accessed on 16 July 2021).
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 2021, 39. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, P.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Chen, S.; Huang, Z.; et al. The Synergistic Effect of PARP Inhibitors and Immune Checkpoint Inhibitors. Clin. Med. Insights Oncol. 2021, 15. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Åvall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.-B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Mehta, M.P.; Wang, D.; Wang, F.; Kleinberg, L.; Brade, A.; Robins, H.I.; Turaka, A.; Leahy, T.; Medina, D.; Xiong, H.; et al. Veliparib in combination with whole brain radiation therapy in patients with brain metastases: Results of a phase 1 study. J. Neurooncol. 2015, 122, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Chabot, P.; Hsia, T.-C.; Ryu, J.-S.; Gorbunova, V.; Belda-Iniesta, C.; Ball, D.; Kio, E.; Mehta, M.; Papp, K.; Qin, Q.; et al. Veliparib in combination with whole-brain radiation therapy for patients with brain metastases from non-small cell lung cancer: Results of a randomized, global, placebo-controlled study. J. Neurooncol. 2017, 131, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.M.; Thompson, P.; Adesina, A.; Li, X.-N.; Kilburn, L.; Onar-Thomas, A.; Kocak, M.; Chyla, B.; McKeegan, E.; Warren, K.E.; et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: A pediatric brain tumor consortium report. Neuro Oncol. 2014, 16, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- Baxter, P.A.; Su, J.M.; Onar-Thomas, A.; Billups, C.A.; Li, X.N.; Poussaint, T.Y.; Smith, E.R.; Thompson, P.; Adesina, A.; Ansell, P.; et al. A phase I/II study of veliparib (ABT-888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: A Pediatric Brain Tumor Consortium study. Neuro Oncol. 2020, 22, 875–885. [Google Scholar] [CrossRef]
- Kleinberg, L.; Supko, J.G.; Mikkelsen, T.; Blakeley, J.O.N.; Stevens, G.; Ye, X.; Desideri, S.; Ryu, S.; Desai, B.; Giranda, V.L.; et al. Phase I adult brain tumor consortium (ABTC) trial of ABT-888 (veliparib), temozolomide (TMZ), and radiotherapy (RT) for newly diagnosed glioblastoma multiforme (GBM) including pharmacokinetic (PK) data. J. Clin. Oncol. 2013, 31, 2065. [Google Scholar] [CrossRef]
- Robins, H.I.; Zhang, P.; Gilbert, M.; Chakravarti, A.; deGroot, J.; Grimm, S.; Wang, F.; Lieberman, F.; Krauze, A.; Sharma, A.; et al. Atct-27nrg oncology/rtog 0929: A randomized phase i/ii study of abt-888in combination with temozolomide in recurrent temozolomide resistant glioblastoma. Neuro Oncol. 2015, 17, v7. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108, djv369. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Christmann, M.; Verbeek, B.; Roos, W.P.; Kaina, B. O6-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim. Biophys. Acta (BBA) Bioenergy Rev. Cancer 2011, 1816, 179–190. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balvers, R.K.; Lamfers, M.L.; Kloezeman, J.J.; Kleijn, A.; Berghauser Pont, L.M.; Dirven, C.M.; Leenstra, S. ABT-888 enhances cytotoxic effects of temozolomide independent of MGMT status in serum free cultured glioma cells. J. Transl. Med. 2015, 13, 74. [Google Scholar] [CrossRef] [Green Version]
- Ho Kim, E.; Sook Song, H.; Hoon Yoo, S.; Yoon, M. Tumor treating fields inhibit glioblastoma cell migration, invasion and angiogenesis. Oncotarget 2016, 7, 65125–65136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galia, A.; Calogero, A.E.; Condorelli, R.; Fraggetta, F.; La Corte, A.; Ridolfo, F.; Bosco, P.; Castiglione, R.; Salemi, M. PARP-1 protein expression in glioblastoma multiforme. Eur. J. Histochem. 2012, 56, e9. [Google Scholar] [CrossRef] [Green Version]
- Piel, M.; Vernaleken, I.; Rösch, F. Positron Emission Tomography in CNS Drug Discovery and Drug Monitoring. J. Med. Chem. 2014, 57, 9232–9258. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, A.; Sharip, A.; Sharip, A.; Jiang, J.; Yang, Q.; Xie, Y. Reverse the Resistance to PARP Inhibitors. Int. J. Biol. Sci. 2017, 13, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Drug/Company | Study Start | Associated Therapy | Clinical Trial | Molecular Biomarker | Status | Cancer Type |
|---|---|---|---|---|---|---|
| Olaparib, OR Lynparza, OR (AZD2281) KuDOS/Astra-Zeneca | Mar 2018 | monotherapy | NCT03212274 Phase II | mutated IDH1/2 | recruiting | glioblastoma |
| Jun 2018 | monotherapy | NCT03561870 Phase II | mutated IDH1/2 | active, not recruiting | recurrent high-grade glioma | |
| Jul 2011 | temozolomide | NCT01390571 Phase I | DNA repair genes, PTEN, MGMT methylation status, mismatch repair | completed | relapsed GBM | |
| Veliparib OR (ABT888) Abbvie | Dec 2018 | radiation therapy and temozolomide | NCT03581292 Phase II | DDR markers | recruiting | glioma without H3 K27M OR BRAFV600E mutations |
| Dec 2014 | temozolomide | NCT02152982 Phase II and III | mutated IDH1/2 MGMT | active, not recruiting | newly diagnosed GBM | |
| Jul 2009 | temozolomide | NCT00946335 Phase I | MGMT, mismatch, DNA repair pathways | completed | pediatric gliomas | |
| Niraparib OR, Zejula, OR (MK4827) Merk/Tesaro | Dec 2019 | tumor treating field | NCT04221503 Phase II | methylation status of MGMT | recruiting | recurrent GBM |
| Pamiparib OR (BGB290) Beigene | Apr 2019 | temozolomide | NCT03749187 Phase I | mutated IDH1/2 and others | recruiting | newly diagnosed glioma grade II OR III |
| Jan 2020 | temozolomide | NCT03914742 Phase I and II | mutated IDH1/2 | recruiting | recurrent GBM | |
| Aug 2017 | radiation therapy and/or temozolomide | NCT03150862 Phase I and II | MGMT Methylation | active, not recruiting | newly diagnosed or recurrent/refractory glioblastoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xavier, M.-A.; Rezende, F.; Titze-de-Almeida, R.; Cornelissen, B. BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme. Biomolecules 2021, 11, 1188. https://doi.org/10.3390/biom11081188
Xavier M-A, Rezende F, Titze-de-Almeida R, Cornelissen B. BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme. Biomolecules. 2021; 11(8):1188. https://doi.org/10.3390/biom11081188
Chicago/Turabian StyleXavier, Mary-Ann, Fernando Rezende, Ricardo Titze-de-Almeida, and Bart Cornelissen. 2021. "BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme" Biomolecules 11, no. 8: 1188. https://doi.org/10.3390/biom11081188
APA StyleXavier, M.-A., Rezende, F., Titze-de-Almeida, R., & Cornelissen, B. (2021). BRCAness as a Biomarker of Susceptibility to PARP Inhibitors in Glioblastoma Multiforme. Biomolecules, 11(8), 1188. https://doi.org/10.3390/biom11081188