
Involvement of Tricarboxylic Acid Cycle Metabolites in Kidney Diseases

 ,
,  ,
, 
Abstract

1. Introduction
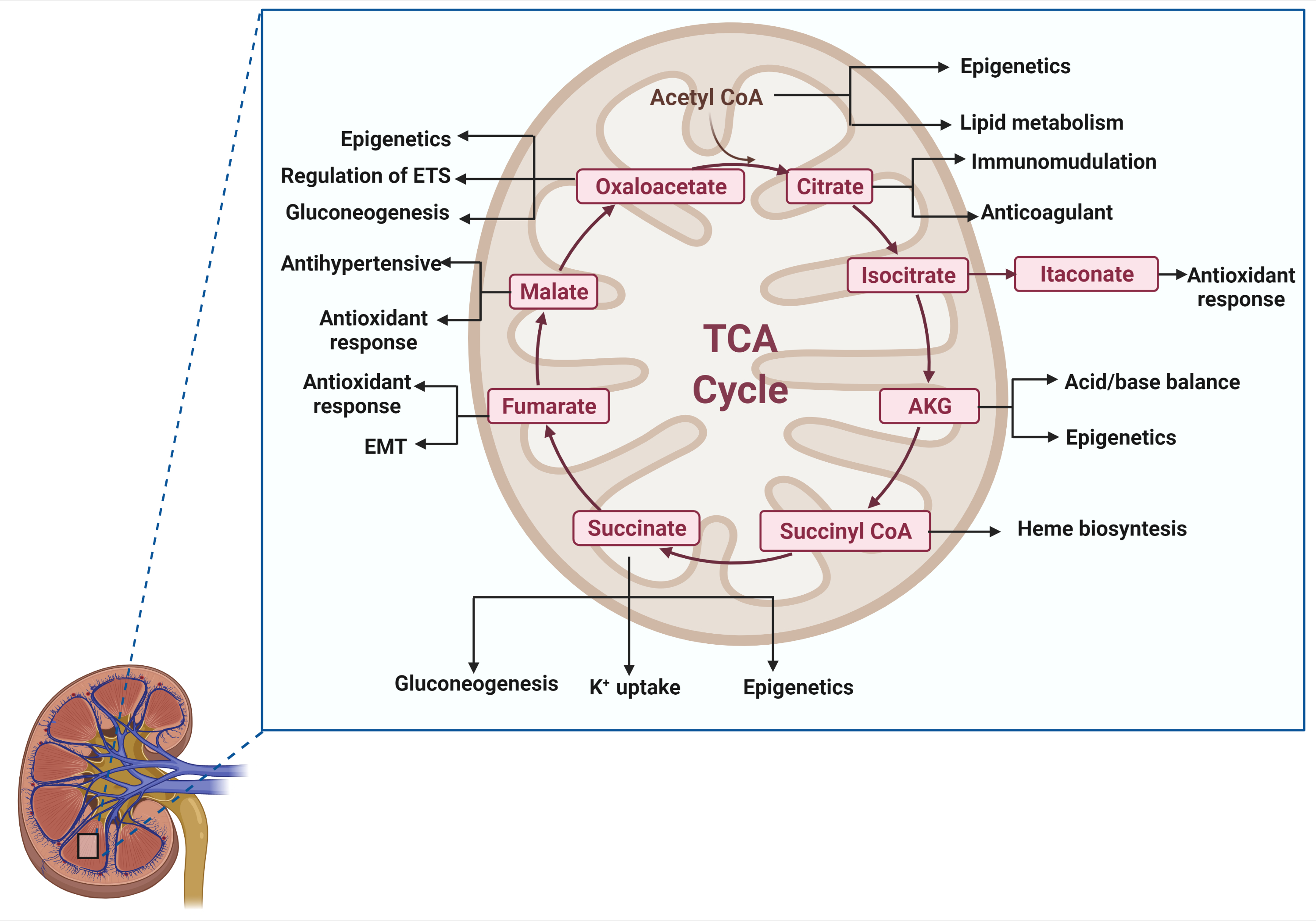
2. A Brief Overview of the TCA Cycle
3. Acetyl-CoA
4. Citrate
5. Isocitrate/Itaconate
6. Alpha-Ketoglutarate
7. Succinyl-CoA
8. Succinate
9. Fumarate
10. Malate
11. Oxaloacetate
12. Clinical Significance of TCA Metabolites
13. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Bononi, A.; Missiroli, S.; Poletti, F.; Suski, J.M.; Agnoletto, C.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Patergnani, S.; et al. Mitochondria-associated membranes (MAMs) as hotspot Ca(2+) signaling units. Adv. Exp. Med. Biol. 2012, 740, 411–437. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, H. Krebs and his trinity of cycles. Nat. Rev. Mol. Cell Biol. 2000, 1, 225–228. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef]
- Afshinnia, F.; Nair, V.; Lin, J.; Rajendiran, T.M.; Soni, T.; Byun, J.; Sharma, K.; Fort, P.E.; Gardner, T.W.; Looker, H.C.; et al. Increased lipogenesis and impaired beta-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Yuan, Q.; Qin, N.; Shi, C.; Luo, J.; Fang, Y.; Xu, L.; Sun, Q.; Zen, K.; Jiang, L.; et al. UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis. 2020, 11, 26. [Google Scholar] [CrossRef]
- Yan, Q.; Song, Y.; Zhang, L.; Chen, Z.; Yang, C.; Liu, S.; Yuan, X.; Gao, H.; Ding, G.; Wang, H. Autophagy activation contributes to lipid accumulation in tubular epithelial cells during kidney fibrosis. Cell Death Discov. 2018, 4, 2. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Y.; Liu, Q.; Wu, F.; Liu, X.; Qu, H.; Yuan, Y.; Ge, J.; Xu, Y.; Wang, H. The mRNA Expression Signature and Prognostic Analysis of Multiple Fatty Acid Metabolic Enzymes in Clear Cell Renal Cell Carcinoma. J. Cancer 2019, 10, 6599–6607. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The Force that Drives Chronic Kidney Disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of hypoxia-inducible factor-2alpha in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Tanada, Y.; Okuda, J.; Kato, T.; Minamino-Muta, E.; Murata, I.; Soga, T.; Shioi, T.; Kimura, T. The metabolic profile of a rat model of chronic kidney disease. PeerJ 2017, 5, e3352. [Google Scholar] [CrossRef]
- Smith, E.R.; Hewitson, T.D. TGF-beta1 is a regulator of the pyruvate dehydrogenase complex in fibroblasts. Sci. Rep. 2020, 10, 17914. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Wigg, B.; Holt, S.; Hewitson, T.D. TGF-beta1 modifies histone acetylation and acetyl-coenzyme A metabolism in renal myofibroblasts. Am. J. Physiol. Renal Physiol. 2019, 316, F517–F529. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Holt, S.G.; Tan, S.J.; Wigg, B.; Samuel, C.S.; Smith, E.R. Epigenetic Modifications to H3K9 in Renal Tubulointerstitial Cells after Unilateral Ureteric Obstruction and TGF-beta1 Stimulation. Front. Pharmacol. 2017, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, S.G.; Gaikwad, A.B.; Lichtnekert, J.; Kulkarni, O.; Eulberg, D.; Klussmann, S.; Tikoo, K.; Anders, H.J. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol. Dial. Transplant. 2010, 25, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Deb, D.K.; Chen, Y.; Sun, J.; Wang, Y.; Li, Y.C. ATP-citrate lyase is essential for high glucose-induced histone hyperacetylation and fibrogenic gene upregulation in mesangial cells. Am. J. Physiol. Renal Physiol. 2017, 313, F423–F429. [Google Scholar] [CrossRef]
- Chen, Y.; Deb, D.K.; Fu, X.; Yi, B.; Liang, Y.; Du, J.; He, L.; Li, Y.C. ATP-citrate lyase is an epigenetic regulator to promote obesity-related kidney injury. FASEB J. 2019, 33, 9602–9615. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Jiang, W.; Yuan, X.; Zhu, H.; He, C.; Ge, C.; Tang, Q.; Xu, C.; Hu, B.; Huang, C.; Ma, T. Inhibition of Histone H3K27 Acetylation Orchestrates Interleukin-9-Mediated and Plays an Anti-Inflammatory Role in Cisplatin-Induced Acute Kidney Injury. Front. Immunol. 2020, 11, 231. [Google Scholar] [CrossRef]
- Zhu, H.; Jiang, W.; Zhao, H.; He, C.; Tang, X.; Xu, S.; Xu, C.; Feng, R.; Li, J.; Ma, T.; et al. PSTPIP2 inhibits cisplatin-induced acute kidney injury by suppressing apoptosis of renal tubular epithelial cells. Cell Death Dis. 2020, 11, 1057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; He, J.; Jia, Z.; Yan, Z.; Yang, J. Acetyl-CoA synthetase 2 enhances tumorigenesis and is indicative of a poor prognosis for patients with renal cell carcinoma. Urol. Oncol. 2018, 36, 243.e9–243.e20. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Guo, X.; Gui, Y. Acetyl-CoA Synthetase 2 Promotes Cell Migration and Invasion of Renal Cell Carcinoma by Upregulating Lysosomal-Associated Membrane Protein 1 Expression. Cell Physiol. Biochem. 2018, 45, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Jiang, L.; Zhang, F.; Li, M.; Yang, B.; Zhang, F.; Guo, X. Acetate promotes SNAI1 expression by ACSS2-mediated histone acetylation under glucose limitation in renal cell carcinoma cell. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Cai, J. Roles of transcriptional factor Snail and adhesion factor E-cadherin in clear cell renal cell carcinoma. Exp. Ther. Med. 2013, 6, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Kanao, K.; Mikami, S.; Mizuno, R.; Shinojima, T.; Murai, M.; Oya, M. Decreased acetylation of histone H3 in renal cell carcinoma: A potential target of histone deacetylase inhibitors. J. Urol. 2008, 180, 1131–1136. [Google Scholar] [CrossRef]
- Usenik, A.; Legisa, M. Evolution of allosteric citrate binding sites on 6-phosphofructo-1-kinase. PLoS ONE 2010, 5, e15447. [Google Scholar] [CrossRef]
- Taylor, W.M.; Halperin, M.L. Regulation of pyruvate dehydrogenase in muscle: Inhibition by citrate. J. Biol. Chem. 1973, 248, 6080–6083. [Google Scholar] [CrossRef]
- MacLellan, D.L.; Mataija, D.; Doucette, A.; Huang, W.; Langlois, C.; Trottier, G.; Burton, I.W.; Walter, J.A.; Karakach, T.K. Alterations in urinary metabolites due to unilateral ureteral obstruction in a rodent model. Mol. Biosyst. 2011, 7, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.; Aa, J.; Qin, W.; Zha, W.; Ge, Y.; Liu, L.; Zheng, T.; Cao, B.; Shi, J.; et al. GC/TOFMS analysis of metabolites in serum and urine reveals metabolic perturbation of TCA cycle in db/db mice involved in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2013, 304, F1317–F1324. [Google Scholar] [CrossRef]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef]
- Goraya, N.; Simoni, J.; Sager, L.N.; Madias, N.E.; Wesson, D.E. Urine citrate excretion as a marker of acid retention in patients with chronic kidney disease without overt metabolic acidosis. Kidney Int. 2019, 95, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Liu, S.; Gurung, R.L.; Ching, J.; Kovalik, J.P.; Tan, T.Y.; Lim, S.C. Urine Tricarboxylic Acid Cycle Metabolites Predict Progressive Chronic Kidney Disease in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 4357–4364. [Google Scholar] [CrossRef]
- Chen, L.; Chen, D.Q.; Liu, J.R.; Zhang, J.; Vaziri, N.D.; Zhuang, S.; Chen, H.; Feng, Y.L.; Guo, Y.; Zhao, Y.Y. Unilateral ureteral obstruction causes gut microbial dysbiosis and metabolome disorders contributing to tubulointerstitial fibrosis. Exp. Mol. Med. 2019, 51, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, W.; He, Q.; Xue, J.; Wang, J.; Xiong, C.; Pu, X.; Nie, Z. Mass Spectrometry Imaging of Kidney Tissue Sections of Rat Subjected to Unilateral Ureteral Obstruction. Sci. Rep. 2017, 7, 41954. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Ullian, M.E.; Robinson, C.J.; Evans, C.T.; Melnick, J.Z.; Fitzgibbon, W.R. Role of citrate synthase in aldosterone-mediated sodium reabsorption. Hypertension 2000, 35, 875–879. [Google Scholar] [CrossRef]
- Minakuchi, H.; Wakino, S.; Urai, H.; Kurokochi, A.; Hasegawa, K.; Kanda, T.; Tokuyama, H.; Itoh, H. The effect of aldosterone and aldosterone blockade on the progression of chronic kidney disease: A randomized placebo-controlled clinical trial. Sci. Rep. 2020, 10, 16626. [Google Scholar] [CrossRef]
- Wei, Q.; Xiao, X.; Fogle, P.; Dong, Z. Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS ONE 2014, 9, e106647. [Google Scholar] [CrossRef]
- Toyohara, T.; Akiyama, Y.; Suzuki, T.; Takeuchi, Y.; Mishima, E.; Tanemoto, M.; Momose, A.; Toki, N.; Sato, H.; Nakayama, M.; et al. Metabolomic profiling of uremic solutes in CKD patients. Hypertens Res. 2010, 33, 944–952. [Google Scholar] [CrossRef]
- Phillips, R.; Hanchanale, V.S.; Myatt, A.; Somani, B.; Nabi, G.; Biyani, C.S. Citrate salts for preventing and treating calcium containing kidney stones in adults. Cochrane Database Syst. Rev. 2015, CD010057. [Google Scholar] [CrossRef]
- Fiaccadori, E.; Regolisti, G.; Cademartiri, C.; Cabassi, A.; Picetti, E.; Barbagallo, M.; Gherli, T.; Castellano, G.; Morabito, S.; Maggiore, U. Efficacy and safety of a citrate-based protocol for sustained low-efficiency dialysis in AKI using standard dialysis equipment. Clin. J. Am. Soc. Nephrol. 2013, 8, 1670–1678. [Google Scholar] [CrossRef]
- Hanevold, C.; Lu, S.; Yonekawa, K. Utility of citrate dialysate in management of acute kidney injury in children. Hemodial. Int. 2010, 14 (Suppl. S1), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Mariano, F.; Bergamo, D.; Gangemi, E.N.; Hollo, Z.; Stella, M.; Triolo, G. Citrate anticoagulation for continuous renal replacement therapy in critically ill patients: Success and limits. Int. J. Nephrol. 2011, 2011, 748320. [Google Scholar] [CrossRef] [PubMed]
- Krieger, N.S.; Asplin, J.R.; Frick, K.K.; Granja, I.; Culbertson, C.D.; Ng, A.; Grynpas, M.D.; Bushinsky, D.A. Effect of Potassium Citrate on Calcium Phosphate Stones in a Model of Hypercalciuria. J. Am. Soc. Nephrol. 2015, 26, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Bienholz, A.; Reis, J.; Sanli, P.; de Groot, H.; Petrat, F.; Guberina, H.; Wilde, B.; Witzke, O.; Saner, F.H.; Kribben, A.; et al. Citrate shows protective effects on cardiovascular and renal function in ischemia-induced acute kidney injury. BMC Nephrol. 2017, 18, 130. [Google Scholar] [CrossRef]
- Tiranathanagul, K.; Jearnsujitwimol, O.; Susantitaphong, P.; Kijkriengkraikul, N.; Leelahavanichkul, A.; Srisawat, N.; Praditpornsilpa, K.; Eiam-Ong, S. Regional citrate anticoagulation reduces polymorphonuclear cell degranulation in critically ill patients treated with continuous venovenous hemofiltration. Ther. Apher. Dial. 2011, 15, 556–564. [Google Scholar] [CrossRef]
- Ou, Y.; Li, S.; Zhu, X.; Gui, B.; Yao, G.; Ma, L.; Zhu, D.; Fu, R.; Ge, H.; Wang, L.; et al. Citrate Attenuates Adenine-Induced Chronic Renal Failure in Rats by Modulating the Th17/Treg Cell Balance. Inflammation 2016, 39, 79–86. [Google Scholar] [CrossRef]
- Choi, E.Y.; Kim, H.J.; Han, J.S. Anti-inflammatory effects of calcium citrate in RAW 264.7cells via suppression of NF-kappaB activation. Environ. Toxicol. Pharmacol. 2015, 39, 27–34. [Google Scholar] [CrossRef]
- Ashbrook, M.J.; McDonough, K.L.; Pituch, J.J.; Christopherson, P.L.; Cornell, T.T.; Selewski, D.T.; Shanley, T.P.; Blatt, N.B. Citrate modulates lipopolysaccharide-induced monocyte inflammatory responses. Clin. Exp. Immunol. 2015, 180, 520–530. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Chen, Y.; Cao, Y.; Wang, W.; Xu, Y.; Wang, Y.; Lv, J.; Li, C.; Su, Y. Overexpression of ATP citrate lyase in renal cell carcinoma tissues and its effect on the human renal carcinoma cells in vitro. Oncol. Lett. 2018, 15, 6967–6974. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014, 19, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Gyuraszova, M.; Gurecka, R.; Babickova, J.; Tothova, L. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxid. Med. Cell Longev. 2020, 2020, 5478708. [Google Scholar] [CrossRef]
- Correa, F.; Buelna-Chontal, M.; Hernandez-Resendiz, S.; Garcia-Nino, W.R.; Roldan, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverri, J.; Tapia, E.; et al. Curcumin maintains cardiac and mitochondrial function in chronic kidney disease. Free Radic. Biol. Med. 2013, 61, 119–129. [Google Scholar] [CrossRef]
- Tapia, E.; Sanchez-Lozada, L.G.; Garcia-Nino, W.R.; Garcia, E.; Cerecedo, A.; Garcia-Arroyo, F.E.; Osorio, H.; Arellano, A.; Cristobal-Garcia, M.; Loredo, M.L.; et al. Curcumin prevents maleate-induced nephrotoxicity: Relation to hemodynamic alterations, oxidative stress, mitochondrial oxygen consumption and activity of respiratory complex I. Free Radic. Res. 2014, 48, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Nilakantan, V.; Liang, H.L.; Rajesh, S.; Mortensen, J.; Chandran, K. Time-dependant protective effects of mangenese(III) tetrakis (1-methyl-4-pyridyl) porphyrin on mitochondrial function following renal ischemia-reperfusion injury. Free Radic. Res. 2010, 44, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Yarian, C.S.; Toroser, D.; Sohal, R.S. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech. Ageing Dev. 2006, 127, 79–84. [Google Scholar] [CrossRef]
- Hooftman, A.; O’Neill, L.A.J. The Immunomodulatory Potential of the Metabolite Itaconate. Trends Immunol. 2019, 40, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zhao, Y.; Luo, Y.; Qian, X.; Zhang, Z.; Jiang, G.; Guo, F. Irg1-itaconate axis protects against acute kidney injury via activation of Nrf2. Am. J. Transl. Res. 2021, 13, 1155–1169. [Google Scholar] [PubMed]
- Tian, F.; Wang, Z.; He, J.; Zhang, Z.; Tan, N. 4-Octyl itaconate protects against renal fibrosis via inhibiting TGF-beta/Smad pathway, autophagy and reducing generation of reactive oxygen species. Eur J. Pharmacol. 2020, 873, 172989. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef]
- Honer Zu Bentrup, K.; Miczak, A.; Swenson, D.L.; Russell, D.G. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J. Bacteriol 1999, 181, 7161–7167. [Google Scholar] [CrossRef]
- Zdzisinska, B.; Zurek, A.; Kandefer-Szerszen, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. 2017, 65, 21–36. [Google Scholar] [CrossRef]
- Otto, C.; Yovkova, V.; Barth, G. Overproduction and secretion of alpha-ketoglutaric acid by microorganisms. Appl. Microbiol. Biotechnol. 2011, 92, 689–695. [Google Scholar] [CrossRef]
- You, Y.H.; Quach, T.; Saito, R.; Pham, J.; Sharma, K. Metabolomics Reveals a Key Role for Fumarate in Mediating the Effects of NADPH Oxidase 4 in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 466–481. [Google Scholar] [CrossRef]
- Salek, R.M.; Maguire, M.L.; Bentley, E.; Rubtsov, D.V.; Hough, T.; Cheeseman, M.; Nunez, D.; Sweatman, B.C.; Haselden, J.N.; Cox, R.D.; et al. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol. Genom. 2007, 29, 99–108. [Google Scholar] [CrossRef]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Xiong, Y.; Peng, T.; Lu, M.; Zhang, L.; Guo, Z. Wild-type IDH1 inhibits the tumor growth through degrading HIF-alpha in renal cell carcinoma. Int. J. Biol. Sci. 2021, 17, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Al-Khallaf, H. Isocitrate dehydrogenases in physiology and cancer: Biochemical and molecular insight. Cell Biosci. 2017, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Jang, H.S.; Noh, M.R.; Kim, J.; Kong, M.J.; Kim, J.I.; Park, J.W.; Park, K.M. Mitochondrial NADP(+)-Dependent Isocitrate Dehydrogenase Deficiency Exacerbates Mitochondrial and Cell Damage after Kidney Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 1200–1215. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.H.; Son, M.K.; Koh, H.J.; Lee, S.M.; Song, I.H.; Kim, Y.O.; Lee, Y.S.; Jeong, K.S.; Kim, W.B.; Park, J.W.; et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J. Biol. Chem. 2001, 276, 16168–16176. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Velvizhi, S.; Nagalashmi, T.; Essa, M.M.; Dakshayani, K.B.; Subramanian, P. Effects of alpha-ketoglutarate on lipid peroxidation and antioxidant status during chronic ethanol administration in Wistar rats. Pol. J. Pharmacol. 2002, 54, 231–236. [Google Scholar]
- Mehra, L.; Hasija, Y.; Mittal, G. Therapeutic potential of alpha-ketoglutarate against acetaminophen-induced hepatotoxicity in rats. J. Pharm. Bioallied. Sci. 2016, 8, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Velvizhi, S.; Dakshayani, K.B.; Subramanian, P. Effects of alpha-ketoglutarate on antioxidants and lipid peroxidation products in rats treated with ammonium acetate. Nutrition 2002, 18, 747–750. [Google Scholar] [CrossRef]
- Kong, M.J.; Han, S.J.; Kim, J.I.; Park, J.W.; Park, K.M. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. 2018, 9, 488. [Google Scholar] [CrossRef]
- Kim, J.; Kim, K.Y.; Jang, H.S.; Yoshida, T.; Tsuchiya, K.; Nitta, K.; Park, J.W.; Bonventre, J.V.; Park, K.M. Role of cytosolic NADP+-dependent isocitrate dehydrogenase in ischemia-reperfusion injury in mouse kidney. Am. J. Physiol. Renal Physiol. 2009, 296, F622–F633. [Google Scholar] [CrossRef]
- Noh, M.R.; Kong, M.J.; Han, S.J.; Kim, J.I.; Park, K.M. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol. 2020, 34, 101548. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Noh, M.R.; Yoon, G.E.; Jang, H.S.; Kong, M.J.; Park, K.M. IDH2 gene deficiency accelerates unilateral ureteral obstruction-induced kidney inflammation through oxidative stress and activation of macrophages. Korean J. Physiol. Pharmacol. 2021, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Laba, P.; Wang, J.; Zhang, J. Low level of isocitrate dehydrogenase 1 predicts unfavorable postoperative outcomes in patients with clear cell renal cell carcinoma. BMC Cancer 2018, 18, 852. [Google Scholar] [CrossRef]
- Tokonami, N.; Morla, L.; Centeno, G.; Mordasini, D.; Ramakrishnan, S.K.; Nikolaeva, S.; Wagner, C.A.; Bonny, O.; Houillier, P.; Doucet, A.; et al. alpha-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J. Clin. Investig. 2013, 123, 3166–3171. [Google Scholar] [CrossRef]
- Weinberg, J.M.; Venkatachalam, M.A.; Roeser, N.F.; Nissim, I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. USA 2000, 97, 2826–2831. [Google Scholar] [CrossRef]
- Bienholz, A.; Petrat, F.; Wenzel, P.; Ickerott, P.; Weinberg, J.M.; Witzke, O.; Kribben, A.; de Groot, H.; Feldkamp, T. Adverse effects of alpha-ketoglutarate/malate in a rat model of acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 303, F56–F63. [Google Scholar] [CrossRef]
- Peralta, C.A.; Hicks, L.S.; Chertow, G.M.; Ayanian, J.Z.; Vittinghoff, E.; Lin, F.; Shlipak, M.G. Control of hypertension in adults with chronic kidney disease in the United States. Hypertension 2005, 45, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Koivunen, P.; Kaelin, W.G., Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 2020, 20, 710–726. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Jaffe, J.; Michael, M.; Swan, C.E.; Duffy, K.J.; Erickson-Miller, C.L.; Haase, V.H. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F1172–F1179. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, X.; Zhang, Y.; Ding, G.; Zhu, C.; Huang, S.; Jia, Z.; Zhang, A. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 825–838. [Google Scholar] [CrossRef]
- Li, X.; Zou, Y.; Xing, J.; Fu, Y.Y.; Wang, K.Y.; Wan, P.Z.; Zhai, X.Y. Pretreatment with Roxadustat (FG-4592) Attenuates Folic Acid-Induced Kidney Injury through Antiferroptosis via Akt/GSK-3beta/Nrf2 Pathway. Oxid. Med. Cell Longev. 2020, 2020, 6286984. [Google Scholar] [CrossRef]
- Kabei, K.; Tateishi, Y.; Nozaki, M.; Tanaka, M.; Shiota, M.; Osada-Oka, M.; Nishide, S.; Uchida, J.; Nakatani, T.; Tomita, S.; et al. Role of hypoxia-inducible factor-1 in the development of renal fibrosis in mouse obstructed kidney: Special references to HIF-1 dependent gene expression of profibrogenic molecules. J. Pharmacol. Sci. 2018, 136, 31–38. [Google Scholar] [CrossRef]
- Del Balzo, U.; Signore, P.E.; Walkinshaw, G.; Seeley, T.W.; Brenner, M.C.; Wang, Q.; Guo, G.; Arend, M.P.; Flippin, L.A.; Chow, F.A.; et al. Nonclinical Characterization of the Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat, a Novel Treatment of Anemia of Chronic Kidney Disease. J. Pharmacol. Exp. Ther. 2020, 374, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Kabei, K.; Tateishi, Y.; Shiota, M.; Osada-Oka, M.; Nishide, S.; Uchida, J.; Nakatani, T.; Matsunaga, S.; Yamaguchi, T.; Tomita, S.; et al. Effects of orally active hypoxia inducible factor alpha prolyl hydroxylase inhibitor, FG4592 on renal fibrogenic potential in mouse unilateral ureteral obstruction model. J. Pharmacol. Sci. 2020, 142, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Brigandi, R.A.; Johnson, B.; Oei, C.; Westerman, M.; Olbina, G.; de Zoysa, J.; Roger, S.D.; Sahay, M.; Cross, N.; McMahon, L.; et al. A Novel Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor (GSK1278863) for Anemia in CKD: A 28-Day, Phase 2A Randomized Trial. Am. J. Kidney Dis. 2016, 67, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Seethy, A.; Pethusamy, K.; Chattopadhyay, I.; Sah, R.; Chopra, A.; Dhar, R.; Karmakar, S. TETology: Epigenetic Mastermind in Action. Appl. Biochem. Biotechnol. 2021, 193, 1701–1726. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, J.; Zhang, H.; Shen, Z.; Liu, H.; Lv, S.; Yu, X.; Zhang, D.; Ding, X.; Zhang, X. Hydrogen sulfide attenuates renal fibrosis by inducing TET-dependent DNA demethylation on Klotho promoter. FASEB J. 2020, 34, 11474–11487. [Google Scholar] [CrossRef]
- Huang, N.; Tan, L.; Xue, Z.; Cang, J.; Wang, H. Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem. Biophys. Res. Commun. 2012, 422, 697–702. [Google Scholar] [CrossRef]
- Yan, H.; Tan, L.; Liu, Y.; Huang, N.; Cang, J.; Wang, H. Ten-eleven translocation methyl-cytosine dioxygenase 2 deficiency exacerbates renal ischemia-reperfusion injury. Clin. Epigenetics 2020, 12, 98. [Google Scholar] [CrossRef]
- Bao, Y.; Bai, M.; Zhu, H.; Yuan, Y.; Wang, Y.; Zhang, Y.; Wang, J.; Xie, X.; Yao, X.; Mao, J.; et al. DNA demethylase Tet2 suppresses cisplatin-induced acute kidney injury. Cell Death Discov. 2021, 7, 167. [Google Scholar] [CrossRef]
- Tampe, B.; Tampe, D.; Zeisberg, E.M.; Muller, G.A.; Bechtel-Walz, W.; Koziolek, M.; Kalluri, R.; Zeisberg, M. Induction of Tet3-dependent Epigenetic Remodeling by Low-dose Hydralazine Attenuates Progression of Chronic Kidney Disease. EBioMedicine 2015, 2, 19–36. [Google Scholar] [CrossRef]
- Tampe, B.; Tampe, D.; Muller, C.A.; Sugimoto, H.; LeBleu, V.; Xu, X.; Muller, G.A.; Zeisberg, E.M.; Kalluri, R.; Zeisberg, M. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J. Am. Soc. Nephrol. 2014, 25, 905–912. [Google Scholar] [CrossRef]
- Yu, C.; Xiong, C.; Tang, J.; Hou, X.; Liu, N.; Bayliss, G.; Zhuang, S. Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFbeta and Notch signaling and preserving PTEN expression. Theranostics 2021, 11, 2706–2721. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, N.; Bhagat, T.D.; Cheville, J.; Lohse, C.; Bhattacharyya, S.; Tischer, A.; Machha, V.; Gordon-Mitchell, S.; Choudhary, G.; Wong, L.F.; et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Investig. 2019, 129, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Harzandi, A.; Lee, S.; Bidkhori, G.; Saha, S.; Hendry, B.M.; Mardinoglu, A.; Shoaie, S.; Sharpe, C.C. Acute kidney injury leading to CKD is associated with a persistence of metabolic dysfunction and hypertriglyceridemia. iScience 2021, 24, 102046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Liu, M.; Xu, Y.; Wu, G. Glycolysis-Related Genes Serve as Potential Prognostic Biomarkers in Clear Cell Renal Cell Carcinoma. Oxid. Med. Cell Longev. 2021, 2021, 6699808. [Google Scholar] [CrossRef] [PubMed]
- Deri, M.T.; Kiss, A.F.; Toth, K.; Paulik, J.; Sarvary, E.; Kobori, L.; Monostory, K. End-stage renal disease reduces the expression of drug-metabolizing cytochrome P450s. Pharmacol. Rep. 2020, 72, 1695–1705. [Google Scholar] [CrossRef]
- Helvig, C.F.; Cuerrier, D.; Hosfield, C.M.; Ireland, B.; Kharebov, A.Z.; Kim, J.W.; Ramjit, N.J.; Ryder, K.; Tabash, S.P.; Herzenberg, A.M.; et al. Dysregulation of renal vitamin D metabolism in the uremic rat. Kidney Int. 2010, 78, 463–472. [Google Scholar] [CrossRef]
- Zhao, L.; Gao, H.; Lian, F.; Liu, X.; Zhao, Y.; Lin, D. (1)H-NMR-based metabonomic analysis of metabolic profiling in diabetic nephropathy rats induced by streptozotocin. Am. J. Physiol. Renal Physiol. 2011, 300, F947–F956. [Google Scholar] [CrossRef]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar] [CrossRef]
- Kamarauskaite, J.; Baniene, R.; Trumbeckas, D.; Strazdauskas, A.; Trumbeckaite, S. Increased Succinate Accumulation Induces ROS Generation in In Vivo Ischemia/Reperfusion-Affected Rat Kidney Mitochondria. Biomed. Res. Int. 2020, 2020, 8855585. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, I.; Taheri, S.; Eroglu, E.; Sener, E.F.; Zararsiz, G.; Uzun, I.; Tufan, E.; Mehmetbeyoglu, E.; Korkmaz Bayramov, K.; Sipahioglu, M.H.; et al. Systemic Succinate, Hypoxia-Inducible Factor-1 Alpha, and IL-1beta Gene Expression in Autosomal Dominant Polycystic Kidney Disease with and without Hypertension. Cardiorenal. Med. 2019, 9, 370–381. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- Gullans, S.R.; Brazy, P.C.; Dennis, V.W.; Mandel, L.J. Interactions between gluconeogenesis and sodium transport in rabbit proximal tubule. Am. J. Physiol. 1984, 246, F859–F869. [Google Scholar] [CrossRef] [PubMed]
- Gullans, S.R.; Kone, B.C.; Avison, M.J.; Giebisch, G. Succinate alters respiration, membrane potential, and intracellular K+ in proximal tubule. Am. J. Physiol. 1988, 255, F1170–F1177. [Google Scholar] [CrossRef] [PubMed]
- Robben, J.H.; Fenton, R.A.; Vargas, S.L.; Schweer, H.; Peti-Peterdi, J.; Deen, P.M.; Milligan, G. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009, 76, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Vargas, S.L.; Toma, I.; Kang, J.J.; Meer, E.J.; Peti-Peterdi, J. Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J. Am. Soc. Nephrol. 2009, 20, 1002–1011. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Won, A.J.; Kim, S.; Kim, Y.G.; Kim, K.B.; Choi, W.S.; Kacew, S.; Kim, K.S.; Jung, J.H.; Lee, B.M.; Kim, S.; et al. Discovery of urinary metabolomic biomarkers for early detection of acute kidney injury. Mol. Biosyst. 2016, 12, 133–144. [Google Scholar] [CrossRef]
- Khattri, R.B.; Thome, T.; Ryan, T.E. Tissue-Specific (1)H-NMR Metabolomic Profiling in Mice with Adenine-Induced Chronic Kidney Disease. Metabolites 2021, 11, 45. [Google Scholar] [CrossRef]
- Molina-Jijon, E.; Tapia, E.; Zazueta, C.; El Hafidi, M.; Zatarain-Barron, Z.L.; Hernandez-Pando, R.; Medina-Campos, O.N.; Zarco-Marquez, G.; Torres, I.; Pedraza-Chaverri, J. Curcumin prevents Cr(VI)-induced renal oxidant damage by a mitochondrial pathway. Free Radic. Biol. Med. 2011, 51, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Niknahad, H.; Heidari, R.; Mohammadzadeh, R.; Ommati, M.M.; Khodaei, F.; Azarpira, N.; Abdoli, N.; Zarei, M.; Asadi, B.; Rasti, M.; et al. Sulfasalazine induces mitochondrial dysfunction and renal injury. Ren. Fail. 2017, 39, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Tamura, Y.; Lanaspa, M.A.; Miyazaki, M.; Suzuki, N.; Sato, W.; Maeshima, Y.; Schreiner, G.F.; Villarreal, F.J.; Johnson, R.J.; et al. Epicatechin limits renal injury by mitochondrial protection in cisplatin nephropathy. Am. J. Physiol. Renal Physiol. 2012, 303, F1264–F1274. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Uribe, A.P.; Bellido, B.; Aparicio-Trejo, O.E.; Tapia, E.; Sanchez-Lozada, L.G.; Hernandez-Santos, J.A.; Fernandez-Valverde, F.; Hernandez-Cruz, E.Y.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Temporal characterization of mitochondrial impairment in the unilateral ureteral obstruction model in rats. Free Radic. Biol. Med. 2021, 172, 358–371. [Google Scholar] [CrossRef]
- Mutsaers, H.A.; Wilmer, M.J.; Reijnders, D.; Jansen, J.; van den Broek, P.H.; Forkink, M.; Schepers, E.; Glorieux, G.; Vanholder, R.; van den Heuvel, L.P.; et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta 2013, 1832, 142–150. [Google Scholar] [CrossRef]
- Beach, T.E.; Prag, H.A.; Pala, L.; Logan, A.; Huang, M.M.; Gruszczyk, A.V.; Martin, J.L.; Mahbubani, K.; Hamed, M.O.; Hosgood, S.A.; et al. Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol. 2020, 36, 101640. [Google Scholar] [CrossRef]
- Mohan, D.; Balasubramanian, E.D.; Ravindran, S.; Kurian, G.A. Renal mitochondria can withstand hypoxic/ischemic injury secondary to renal failure in uremic rats pretreated with sodium thiosulfate. Indian J. Pharmacol. 2017, 49, 317–321. [Google Scholar] [CrossRef]
- Sun, G.; Zhang, X.; Liang, J.; Pan, X.; Zhu, S.; Liu, Z.; Armstrong, C.M.; Chen, J.; Lin, W.; Liao, B.; et al. Integrated Molecular Characterization of Fumarate Hydratase-deficient Renal Cell Carcinoma. Clin. Cancer Res. 2021, 27, 1734–1743. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Takasu, C.; Vaziri, N.D.; Li, S.; Robles, L.; Vo, K.; Takasu, M.; Pham, C.; Liu, S.; Farzaneh, S.H.; Foster, C.E., 3rd; et al. Treatment With Dimethyl Fumarate Attenuates Calcineurin Inhibitor-induced Nephrotoxicity. Transplantation 2015, 99, 1144–1150. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, F.; Zhou, N.; Liu, S.; Wang, P.; Zhang, S.; Zhang, Y.; Zhang, A.; Jia, Z.; Huang, S. Dimethyl fumarate prevents ferroptosis to attenuate acute kidney injury by acting on NRF2. Clin. Transl. Med. 2021, 11, e382. [Google Scholar] [CrossRef]
- Sasaki, A.; Koike, N.; Murakami, T.; Suzuki, K. Dimethyl fumarate ameliorates cisplatin-induced renal tubulointerstitial lesions. J. Toxicol. Pathol. 2019, 32, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, C.; Carter, J.L. An evaluation of dimethyl fumarate for the treatment of relapsing remitting multiple sclerosis. Expert. Opin. Pharmacother. 2020, 21, 1399–1405. [Google Scholar] [CrossRef]
- Nielsen, P.M.; Eldirdiri, A.; Bertelsen, L.B.; Jorgensen, H.S.; Ardenkjaer-Larsen, J.H.; Laustsen, C. Fumarase activity: An in vivo and in vitro biomarker for acute kidney injury. Sci. Rep. 2017, 7, 40812. [Google Scholar] [CrossRef]
- Hou, E.; Sun, N.; Zhang, F.; Zhao, C.; Usa, K.; Liang, M.; Tian, Z. Malate and Aspartate Increase L-Arginine and Nitric Oxide and Attenuate Hypertension. Cell Rep. 2017, 19, 1631–1639. [Google Scholar] [CrossRef]
- Bradshaw, P.C. Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging. Nutrients 2019, 11, 504. [Google Scholar] [CrossRef]
- Al Kadhi, O.; Melchini, A.; Mithen, R.; Saha, S. Development of a LC-MS/MS Method for the Simultaneous Detection of Tricarboxylic Acid Cycle Intermediates in a Range of Biological Matrices. J. Anal. Methods Chem. 2017, 2017, 5391832. [Google Scholar] [CrossRef]
- Shil, K.; Pal, S. Metabolic adaptability in hexavalent chromium-treated renal tissue: An in vivo study. Clin. Kidney J. 2018, 11, 222–229. [Google Scholar] [CrossRef]
- Khan, S.A.; Priyamvada, S.; Farooq, N.; Khan, S.; Khan, M.W.; Yusufi, A.N. Protective effect of green tea extract on gentamicin-induced nephrotoxicity and oxidative damage in rat kidney. Pharmacol. Res. 2009, 59, 254–262. [Google Scholar] [CrossRef]
- Rony, K.A.; Ajith, T.A.; Kuttikadan, T.A.; Blaze, R.; Janardhanan, K.K. Phellinus rimosus improves mitochondrial energy status and attenuates nephrotoxicity in diabetic rats. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Sette, L.H.; Lopes, E.P. The reduction of serum aminotransferase levels is proportional to the decline of the glomerular filtration rate in patients with chronic kidney disease. Clinics 2015, 70, 346–349. [Google Scholar] [CrossRef]
- Pandey, N.; Lanke, V.; Vinod, P.K. Network-based metabolic characterization of renal cell carcinoma. Sci. Rep. 2020, 10, 5955. [Google Scholar] [CrossRef] [PubMed]
- Fink, B.D.; Bai, F.; Yu, L.; Sheldon, R.D.; Sharma, A.; Taylor, E.B.; Sivitz, W.I. Oxaloacetic acid mediates ADP-dependent inhibition of mitochondrial complex II-driven respiration. J. Biol. Chem. 2018, 293, 19932–19941. [Google Scholar] [CrossRef]
- Mischak, H.; Delles, C.; Vlahou, A.; Vanholder, R. Proteomic biomarkers in kidney disease: Issues in development and implementation. Nat. Rev. Nephrol. 2015, 11, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Sirolli, V.; Pieroni, L.; Di Liberato, L.; Urbani, A.; Bonomini, M. Urinary Peptidomic Biomarkers in Kidney Diseases. Int. J. Mol. Sci. 2019, 21, 96. [Google Scholar] [CrossRef] [PubMed]
- Dubin, R.F.; Rhee, E.P. Proteomics and Metabolomics in Kidney Disease, including Insights into Etiology, Treatment, and Prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef]
- Inker, L.A.; Okparavero, A. Cystatin C as a marker of glomerular filtration rate: Prospects and limitations. Curr. Opin Nephrol. Hypertens 2011, 20, 631–639. [Google Scholar] [CrossRef]
- Peralta, C.A.; Katz, R.; Sarnak, M.J.; Ix, J.; Fried, L.F.; De Boer, I.; Palmas, W.; Siscovick, D.; Levey, A.S.; Shlipak, M.G. Cystatin C identifies chronic kidney disease patients at higher risk for complications. J. Am. Soc. Nephrol. 2011, 22, 147–155. [Google Scholar] [CrossRef]
- Nielsen, S.E.; Schjoedt, K.J.; Astrup, A.S.; Tarnow, L.; Lajer, M.; Hansen, P.R.; Parving, H.H.; Rossing, P. Neutrophil Gelatinase-Associated Lipocalin (NGAL) and Kidney Injury Molecule 1 (KIM1) in patients with diabetic nephropathy: A cross-sectional study and the effects of lisinopril. Diabet. Med. 2010, 27, 1144–1150. [Google Scholar] [CrossRef]
- Devarajan, P. Proteomics for biomarker discovery in acute kidney injury. Semin. Nephrol. 2007, 27, 637–651. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malyszko, J.; Bachorzewska-Gajewska, H.; Sitniewska, E.; Malyszko, J.S.; Poniatowski, B.; Dobrzycki, S. Serum neutrophil gelatinase-associated lipocalin as a marker of renal function in non-diabetic patients with stage 2-4 chronic kidney disease. Ren. Fail. 2008, 30, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Miyazaki, T.; Honda, A.; Shimohata, H.; Hirayama, K.; Kobayashi, M. Retention of acetylcarnitine in chronic kidney disease causes insulin resistance in skeletal muscle. J. Clin. Biochem. Nutr. 2016, 59, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Q.; Cao, G.; Chen, H.; Argyopoulos, C.P.; Yu, H.; Su, W.; Chen, L.; Samuels, D.C.; Zhuang, S.; Bayliss, G.P.; et al. Identification of serum metabolites associating with chronic kidney disease progression and anti-fibrotic effect of 5-methoxytryptophan. Nat. Commun. 2019, 10, 1476. [Google Scholar] [CrossRef]
- Sun, J.; Shannon, M.; Ando, Y.; Schnackenberg, L.K.; Khan, N.A.; Portilla, D.; Beger, R.D. Serum metabolomic profiles from patients with acute kidney injury: A pilot study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 893–894, 107–113. [Google Scholar] [CrossRef]
- Jing, L.; Guigonis, J.M.; Borchiellini, D.; Durand, M.; Pourcher, T.; Ambrosetti, D. LC-MS based metabolomic profiling for renal cell carcinoma histologic subtypes. Sci. Rep. 2019, 9, 15635. [Google Scholar] [CrossRef]
- Lu, Y.; Li, N.; Gao, L.; Xu, Y.J.; Huang, C.; Yu, K.; Ling, Q.; Cheng, Q.; Chen, S.; Zhu, M.; et al. Acetylcarnitine Is a Candidate Diagnostic and Prognostic Biomarker of Hepatocellular Carcinoma. Cancer Res. 2016, 76, 2912–2920. [Google Scholar] [CrossRef]
- Takaya, H.; Namisaki, T.; Kitade, M.; Shimozato, N.; Kaji, K.; Tsuji, Y.; Nakanishi, K.; Noguchi, R.; Fujinaga, Y.; Sawada, Y.; et al. Acylcarnitine: Useful biomarker for early diagnosis of hepatocellular carcinoma in non-steatohepatitis patients. World J. Gastrointest. Oncol. 2019, 11, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.Y.; Kwon, Y.K.; Lee, D.; Jung, H.Y.; Ryu, H.M.; Cho, J.H.; Ryu, D.H.; Kim, Y.L.; Hwang, G.S. Changes in serum metabolites with the stage of chronic kidney disease: Comparison of diabetes and non-diabetes. Clin. Chim. Acta 2016, 459, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, B.; Fitzpatrick, M.; Al-Nuaimi, H.; Coughlan, A.M.; Hickey, F.B.; Pusey, C.D.; Savage, C.; Benton, C.M.; O’Brien, E.C.; O’Toole, D.; et al. Changes in urinary metabolomic profile during relapsing renal vasculitis. Sci. Rep. 2016, 6, 38074. [Google Scholar] [CrossRef] [PubMed]
- Muhle-Goll, C.; Eisenmann, P.; Luy, B.; Kolker, S.; Tonshoff, B.; Fichtner, A.; Westhoff, J.H. Urinary NMR Profiling in Pediatric Acute Kidney Injury-A Pilot Study. Int. J. Mol. Sci. 2020, 21, 1187. [Google Scholar] [CrossRef]
- Falegan, O.S.; Arnold Egloff, S.A.; Zijlstra, A.; Hyndman, M.E.; Vogel, H.J. Urinary Metabolomics Validates Metabolic Differentiation Between Renal Cell Carcinoma Stages and Reveals a Unique Metabolic Profile for Oncocytomas. Metabolites 2019, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Gisewhite, S.; Stewart, I.J.; Beilman, G.; Lusczek, E. Urinary metabolites predict mortality or need for renal replacement therapy after combat injury. Crit. Care 2021, 25, 119. [Google Scholar] [CrossRef] [PubMed]
- Falegan, O.S.; Ball, M.W.; Shaykhutdinov, R.A.; Pieroraio, P.M.; Farshidfar, F.; Vogel, H.J.; Allaf, M.E.; Hyndman, M.E. Urine and Serum Metabolomics Analyses May Distinguish between Stages of Renal Cell Carcinoma. Metabolites 2017, 7, 6. [Google Scholar] [CrossRef]
- Zheng, P.; Wang, Y.; Chen, L.; Yang, D.; Meng, H.; Zhou, D.; Zhong, J.; Lei, Y.; Melgiri, N.D.; Xie, P. Identification and validation of urinary metabolite biomarkers for major depressive disorder. Mol. Cell Proteom. 2013, 12, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.R.; Coresh, J.; Inker, L.A.; Levey, A.S.; Zheng, Z.; Rebholz, C.M.; Tin, A.; Appel, L.J.; Chen, J.; Sarnak, M.J.; et al. Serum metabolites are associated with all-cause mortality in chronic kidney disease. Kidney Int. 2018, 94, 381–389. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Metabolite | Kidney Disease | References | ||
|---|---|---|---|---|
| CKD | AKI | RCC | ||
| Acetyl-carnitine | Δ serum ∇ urine | Δ serum | Δ tissue | [166,167,168,169] |
| Citrate | ∇ urine | ∇ urine | ∇ urine | [43,62,174,175] |
| Isocitrate | ∇ urine | - | - | [42] |
| AKG | ∇ urine | - | ∇ urine Δ tissue | [42,81,177] |
| Succinate | ∇ urine | - | ∇ urine Δ tissue | [42,62,175] |
| Fumarate | Δ urine | - | ∇ tissue | [44,62] |
| Malate | Δ urine | - | ∇ tissue | [44,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Uribe, A.P.; Hernández-Cruz, E.Y.; Ramírez-Magaña, K.J.; Pedraza-Chaverri, J. Involvement of Tricarboxylic Acid Cycle Metabolites in Kidney Diseases. Biomolecules 2021, 11, 1259. https://doi.org/10.3390/biom11091259
Jiménez-Uribe AP, Hernández-Cruz EY, Ramírez-Magaña KJ, Pedraza-Chaverri J. Involvement of Tricarboxylic Acid Cycle Metabolites in Kidney Diseases. Biomolecules. 2021; 11(9):1259. https://doi.org/10.3390/biom11091259
Chicago/Turabian StyleJiménez-Uribe, Alexis Paulina, Estefani Yaquelin Hernández-Cruz, Karla Jaqueline Ramírez-Magaña, and José Pedraza-Chaverri. 2021. "Involvement of Tricarboxylic Acid Cycle Metabolites in Kidney Diseases" Biomolecules 11, no. 9: 1259. https://doi.org/10.3390/biom11091259
APA StyleJiménez-Uribe, A. P., Hernández-Cruz, E. Y., Ramírez-Magaña, K. J., & Pedraza-Chaverri, J. (2021). Involvement of Tricarboxylic Acid Cycle Metabolites in Kidney Diseases. Biomolecules, 11(9), 1259. https://doi.org/10.3390/biom11091259