
Looking at Alzheimer’s Disease Pathogenesis from the Nuclear Side

Abstract
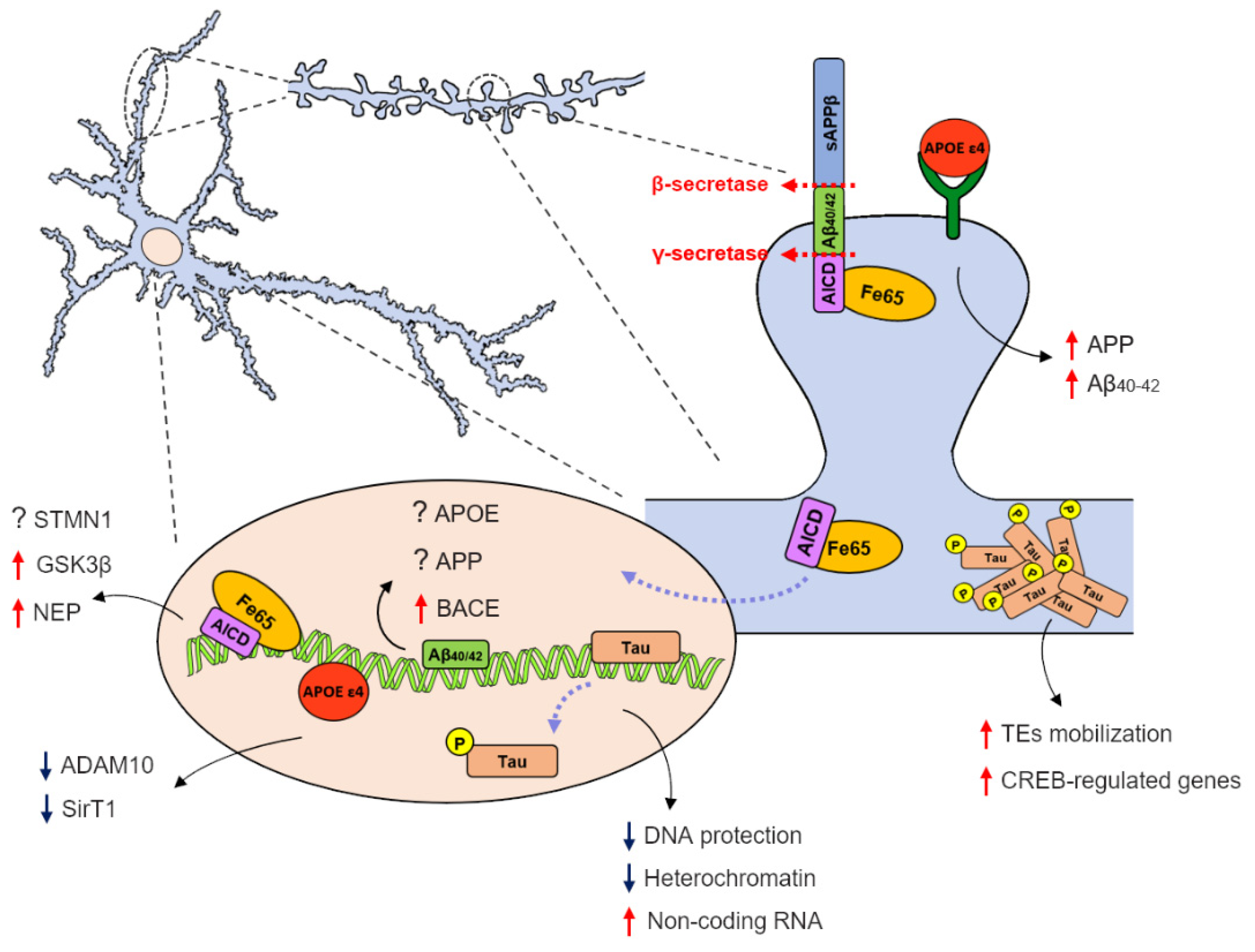
:1. Introduction
2. APP Metabolites and Their Role as Transcription Regulators
2.1. APP Intracellular Domain as a Potential Transcription Factor
2.2. Amyloid-β Nuclear Localization and DNA Binding Properties
3. The Relevance of Nuclear Tau in Alzheimer’s Disease Pathogenesis
4. APOE ε4 as a Transcriptional Regulator
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patterson, C. Alzheimer’s Disease International World Alzheimer Report 2018–The State of the Art of Dementia Research; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004457. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Marcello, E.; Epis, R.; Gardoni, F.; Di Luca, M. The amyloid cascade: The old and the new. J. Nutr. Health Aging 2008, 12, 58S–60S. [Google Scholar] [CrossRef]
- Haass, C.; Hung, A.Y.; Schlossmacher, M.G.; Teplow, D.B.; Selkoe, D.J. β-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 1993, 268, 3021–3024. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Blenkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashler, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β- secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccaveffo, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Voytyuk, I.; De Strooper, B.; Chávez-Gutiérrez, L. Modulation of γ- and β-Secretases as Early Prevention Against Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 320–327. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human g-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, E.; Borroni, B.; Pelucchi, S.; Gardoni, F.; Di Luca, M. ADAM10 as a therapeutic target for brain diseases: From developmental disorders to Alzheimer’s disease. Expert Opin. Ther. Targets 2017, 21, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Pontrello, C.G.; Sun, M.Y.; Lin, A.; Fiacco, T.A.; DeFea, K.A.; Ethell, I.M. Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc. Natl. Acad. Sci. USA 2012, 109, E442–E451. [Google Scholar] [CrossRef] [Green Version]
- Terrill-Usery, S.E.; Colvin, B.A.; Davenport, R.E.; Nichols, M.R. Aβ40 has a subtle effect on Aβ42 protofibril formation, but to a lesser degree than Aβ42 concentration, in Aβ42/Aβ40 mixtures. Arch. Biochem. Biophys. 2016, 597, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A Specific Amyloid-beta Protein Assembly in the Brain Impairs Memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef]
- Iqbal, K.; Zaidi, T.; Wen, G.Y.; Grundke-Iqbal, I.; Merz, P.A.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective brain microtuble assembly in Alzheimer’s disease. Lancet 1986, 2, 421–426. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Saraceno, C.; Musardo, S.; Marcello, E.; Pelucchi, S.; Di Luca, M. Modeling Alzheimer’s disease: From past to future. Front. Pharmacol. 2013, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Epis, R.; Gardoni, F.; Marcello, E.; Genazzani, A.; Canonico, P.L.; Di Luca, M. Searching for new animal models of Alzheimer′s disease. Eur. J. Pharmacol. 2010, 626, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebeck, W.G.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Betensky, R.A.; Hyman, B.T. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann. Neurol. 2015, 77, 917–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.M.; Scheltens, P.; Visser, P.J.; Verfaillie, S.C.J.; Zwan, M.D.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein e, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Hashimoto, T.; Nomoto, H.; Hyman, B.T.; Iwatsubo, T. Role of apolipoprotein E in β-amyloidogenesis: Isoform-specific effects on protofibril to fibril conversion of Aβ in vitro and brain Aβ deposition in vivo. J. Biol. Chem. 2015, 290, 15163–15174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human APOE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Haass, C.; Steiner, H. Regulated intramembrane proteolysis–Lessons from amyloid precursor protein processing. J. Neurochem. 2011, 117, 779–796. [Google Scholar] [CrossRef]
- Selkoe, D.; Kopan, R. Notch and Presenilin: Regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 2003, 26, 565–597. [Google Scholar] [CrossRef]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef]
- Cao, X.; Sudhof, T.C. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef] [Green Version]
- von Rotz, R.C.; Kohli, B.M.; Bosset, J.; Meier, M.; Suzuki, T.; Nitsch, R.M.; Konietzko, U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 2004, 117, 4435–4448. [Google Scholar] [CrossRef] [Green Version]
- Edbauer, D.; Willem, M.; Lammich, S.; Steiner, H.; Haass, C. Insulin-degrading enzyme rapidly removes the β-amyloid precursor protein intracellular domain (AICD). J. Biol. Chem. 2002, 277, 13389–13393. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.C.; Rabizadeh, S.; Chandra, S.; Shayya, R.F.; Ellerby, L.M.; Ye, X.; Salvesen, G.S.; Koo, E.H.; Bredesen, D.E. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat. Med. 2000, 6, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Cupers, P.; Orlans, I.; Craessaerts, K.; Annaert, W.; De Strooper, B. The amyloid precursor protein (APP) -cytoplasmic fragment generated by gamma-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J. Neurochem. 2001, 78, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; Zheng, J.B.; Gue, S.Y.; Selkoe, D.J. The Intracellular Domain of the betaamyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like Manner. J. Biol Chem. 2001. [Google Scholar] [CrossRef] [Green Version]
- Fiore, F.; Zambrano, N.; Minopoli, G.; Donini, V.; Duilio, A.; Russo, T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer’s amyloid precursor protein. J. Biol. Chem. 1995. [Google Scholar] [CrossRef] [Green Version]
- Bressler, S.L.; Gray, M.D.; Sopher, B.L.; Hu, Q.; Hearn, M.G.; Pham, D.G.; Dinulos, M.B.; Fukuchi, K.I.; Sisodia, S.S.; Miller, M.A.; et al. cDNA cloning and chromosome mapping of the human Fe65 gene: Interaction of the conserved cytoplasmic domains of the human β-amyloid precursor protein and its homologues with the mouse Fe65 protein. Hum. Mol. Genet. 1996, 5, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Minopoli, G.; De Candia, P.; Bonetti, A.; Faraonio, R.; Zambrano, N.; Russo, T. The beta-amyloid precursor protein functions as a cytosolic anchoring site that prevents Fe65 nuclear translocation. J. Biol. Chem. 2001, 276, 6545–6550. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Iijima, K.I.; Elliott, J.I.; Kirino, Y.; Suzuki, T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J. Biol. Chem. 2001, 276, 40353–40361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkinton, M.S.; Standen, C.L.; Lau, K.F.; Kesavapany, S.; Byers, H.L.; Ward, M.; McLoughlin, D.M.; Miller, C.C.J. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J. Biol. Chem. 2004, 279, 22084–22091. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, A.; Whelan, C.M.; Smith, C.J.; Berezovska, O.; Hyman, B.T. Direct visualization of the gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein: Association with Fe65 and translocation to the nucleus. J. Neurochem. 2002, 82, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, V.; Kins, S. Fe65: A Scaffolding Protein of Actin Regulators. Cells 2021, 25, 1599. [Google Scholar] [CrossRef]
- Nensa, F.M.; Neumann, M.H.D.; Schrotter, A.; Przyborski, A.; Mastalski, T.; Susdalzew, S.; Looβe, C.; Helling, S.; El Magraoui, F.; Erdmann, R.; et al. Amyloid beta a4 precursor protein-binding family b member 1 (FE65) interactomics revealed synaptic vesicle glycoprotein 2a (SV2A) and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) as new binding proteins in the human brain. Mol. Cell. Proteom. 2014, 13, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Ohgi, K.A.; Rose, D.W.; Koo, E.H.; Glass, C.K.; Rosenfeld, M.G. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-Amyloid Precursor Protein. Cell. 2002, 110, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Schrötter, A.; Loosse, C.; Pfeiffer, K.; Theiss, C.; Kauth, M.; Meyer, H.E.; Marcus, K. A ternary complex consisting of AICD, FE65, and TIP60 down-regulates Stathmin1. Biochim. Biophys. Acta 2013, 1834, 387–394. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Nalivaeva, N.N.; Makova, N.Z.; Turner, A.J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: Implications for Alzheimer disease. EMBO Rep. 2009, 10, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Pardossi-piquard, R.; Petit, A.; Kawarai, T.; Sunyach, C.; Alves, C.; Vincent, B.; Ring, S.; Adamio, L.D.; Shen, J.; Müller, U.; et al. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of beta APP and APLP. Neuron. 2005, 46, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.W.; Mett, J.; Stahlmann, C.P.; Grösgen, S.; Haupenthal, V.J.; Blümel, T.; Hundsdörfer, B.; Zimmer, V.C.; Mylonas, N.T.; Tanila, H.; et al. APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 2015, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.W.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Zimmer, V.C.; Hartmann, T. Neprilysin and Aβ clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front. Aging Neurosci. 2013, 23, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Kim, E.; Lee, J.; Park, C.H.; Kim, S. C-terminal fragments of amyloid precursor protein exert neurotossicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 2003, 17, 1951–1953. [Google Scholar] [CrossRef]
- Ozaki, T.; Li, Y.; Kikuchi, H.; Tomita, T.; Iwatsubo, T.; Nakagawara, A. The intracellular domain of the amyloid precursor protein (AICD) enhances the p53-mediated apoptosis. Biochem. Biophys. Res. Commun. 2006, 351, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Kim, H.; Ha, T.; Ha, J.; Shin, K.Y.; Jeong, Y.H.; Lee, J.; Park, C.; Kim, S.; Baik, T.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell. Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hsu, W.; Ma, Y.; Lee, E.H.Y. Melatonin Induction of APP Intracellular Domain 50 SUMOylation Alleviates AD through Enhanced Transcriptional Activation and AβDegradation. Mol. Ther. 2021, 29, 376–395. [Google Scholar] [CrossRef]
- Pastorino, L.; Sun, A.; Lu, P.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.; Li, X.; et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 2006, 440, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.A.; Pimplikar, S.W. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J. Cell Biol. 2005, 171, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer’s disease. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Gong, K.; Van Laar, T.; Gong, Y.; Zhang, L. Wnt /β-catenin signal pathway stabilizes APP intracellular domain (AICD) and promotes its transcriptional activity. Biochem. Biophys. Res. Commun. 2011, 412, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Piña-crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef]
- Zhou, F.; Gong, K.; Song, B.; Ma, T.; Van Laar, T.; Gong, Y.; Zhang, L. The APP intracellular domain (AICD) inhibits Wnt signalling and promotes neurite outgrowth. Biochim. Biophys. Acta 2012, 1823, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Stathopoulos, A.; Pimplikar, S.W. APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS ONE 2010, 5, e11866. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Fan, Q.; Dawson, H.N.; Pimplikar, S.W. Tau Protein Mediates APP Intracellular Domain (AICD) -Induced Alzheimer ’s-Like Pathological Features in Mice. PLoS ONE 2016, 11, e0159435. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Futagawa, T.; Yang, W.; Jiang, X.; Zeng, L.; Takeda, Y.; Xu, X.; Bagnard, D.; Schachner, M.; Furley, A.J.; et al. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat. Cell. Biol. 2008, 10, 283–294. [Google Scholar] [CrossRef]
- Coronel, R.; Lachgar, M.; Bernabeu-zornoza, A.; Palmer, C.; Domínguez-alvaro, M.; Revilla, A.; Ocaña, I.; Fernández, A.; Martínez-serrano, A.; Cano, E.; et al. Neuronal and Glial Differentiation of Human Neural Stem Cells Is Regulated by Amyloid Precursor Protein (APP) Levels. Mol. Neurobiol. 2019, 56, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Wong, W.; Ma, Q.H.; Yang, Z.Z.; Zhu, H.; Liu, F.J.; Wang, P.; Ma, J.; Yan, S.; Polo, J.M.; et al. APP intracellular domain acts as a transcriptional regulator of miR-663 suppressing neuronal differentiation. Cell Death Dis. 2015, 6, e1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Vanan, S.; Tu, H.; Zhang, W.; Zhang, Z.; Chia, S.; Eun, S.; Zeng, X.; Yu, W.; Xu, J.; et al. Amyloid precursor protein intracellular domain-dependent regulation of FOXO3a inhibits adult hippocampal neurogenesis. Neurobiol. Aging 2020, 95, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Law, B.M.; Guest, A.L.; Pullen, M.W.J.; Perkinton, M.S. Increased Foxo3a Nuclear Translocation and Activity is an Early Neuronal Response to βγ-Secretase-Mediated Processing of the Amyloid-β Protein Precursor: Utility of an APP-GAL4 Reporter Assay. J. Alzheimer’s Dis. 2018, 61, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Goiran, T.; Duplan, E.; Chami, M.; Bourgeois, A.; El Manaa, W.; Rouland, L.; Dunys, J.; Lauritzen, I.; You, H.; Stambolic, V.; et al. β-Amyloid Precursor Protein Intracellular Domain Controls Mitochondrial Function by Modulating Phosphatase and Tensin Homolog—Induced Kinase 1 Transcription in Cells and in Alzheimer Mice Models. Biol. Psychiatry 2018, 83, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Maloney, B.; Lahiri, D.K. The Alzheimer’s amyloid β -peptide (Aβ) binds a specific DNA Aβ-interacting domain (AβID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: Characterizing a new regulatory motif. Gene 2011, 488, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gezen, D.; İrem, A.; Esin, L.A.; Merve, C.; Erdinç, A. The Transcriptional Regulatory Properties of Amyloid Beta 1– 42 may Include Regulation of Genes Related to Neurodegeneration. Neuromol. Med. 2018, 20, 363–375. [Google Scholar] [CrossRef]
- Bailey, J.A.; Maloney, B.; Ge, Y.; Lahiri, D.K. Functional activity of the novel Alzheimer’s amyloid β-peptide interacting domain (Aβ ID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis. Gene 2011, 488, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barucker, C.; Harmeier, A.; Weiske, J.; Fauler, B.; Albring, K.F.; Prokop, S.; Hildebrand, P.; Lurz, R.; Heppner, F.L.; Huber, O.; et al. Nuclear Translocation Uncovers the Amyloid Peptide Aβ42 as a Regulator of Gene Transcription. J. Biol. Chem. 2014, 289, 20182–20191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barucker, C.; Sommer, A.; Beckmann, G.; Eravci, M.; Harmeier, A. Alzheimer Amyloid Peptide aβ42 Regulates Gene Expression of Transcription and Growth Factors. J. Alzheimers Dis. 2015, 44, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Gezen, D. Vitamin D Receptor Regulates Amyloid Beta 1−42 Production with Protein Disulfide Isomerase A3. ACS Chem. Neurosci. 2017, 8, 2335–2346. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Buée, L.; Bussiére, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Maina, M.B.; Al-hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Götz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.A.; Johonson, G.V. Localization and in situ phosphorylation state of nuclear tau. Exp. Cell Res. 1995, 220, 332–337. [Google Scholar] [CrossRef]
- Loomis, P.A.; Howardt, T.H.; Castleberryt, R.P.; Binder, L. 1 Identification of nuclear X isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1990, 87, 8422–8426. [Google Scholar] [CrossRef] [Green Version]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [Green Version]
- Brady, R.M.; Zinkowski, R.P.; Binder, L.I. Presence of tau in isolated nuclei from human brain. Neurobiol. Aging 1995, 16, 479–486. [Google Scholar] [CrossRef]
- Thurston, V.C.; Zikowski, R.P.; Binder, L.I. Tau as a nucleolar protein in human nonneural cells in vitro and in vivo. Chromosoma 1996, 105, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, M.K.; Shestakova, E.; Mansoroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 19, 2025–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiche, G.; Corces, V.G.; Avila, J. Preferential binding of hog brain microtubule-associated proteins to mouse satellite versus bulk DNA preparations. Nature 1978, 273, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Corces, V.G.; Salas, J.; Salas, M.L.; Avila, J. Binding of microtubule proteins to DNA: Specificity of the interaction. Eur. J. Biochem. 1978, 86, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R.Q.; Haque, N.; Qu, M.H.; del Carmen Alonso, A.I.; Grundke-Iqbal, I.; Iqbal, K. Microtubule associated protein tau binds to double-stranded but not single-stranded DNA. Cell. Mol. Life Sci. 2003, 60, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Qu, M.H.; Wang, X.S.; Chen, L.; Wang, D.L.; Liu, Y.; Hua, Q.; He, R.Q. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE 2008, 3, e2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camero, S.; Benítez, M.J.; Barrantes, A.; Ayuso, J.M.; Cuadros, R.; Avila, J.; Jiménez, J.S. Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J. Alzheimer’s Dis. 2014, 39, 649–660. [Google Scholar] [CrossRef]
- Multhaup, G.; Huber, O.; Galas, M. Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Aβ42, and Tau in Nuclear Roles. J. Biolog. Chem. 2015, 290, 23515–23522. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Cantrelle, F.X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finc, C.E.; St Laurent, G.; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Klein, H.U.; McCabe, C.; Gjonesca, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef]
- Nativio, R.; Lan, Y.; Donauhe, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef]
- Olson, M.O.J. Nucleolus: Structure and Function. Encycl. Life Sci. 2006. [Google Scholar] [CrossRef]
- Lu, J.; Miao, J.; Su, T.; Liu, Y.; He, R. Formaldehyde induces hyperphosphorylation and polymerization of Tau protein both in vitro and in vivo. Biochim. Biophys. Acta. 2013, 1830, 4102–4116. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell. Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef]
- Sun, W.; Samini, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Jhonson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Mahoney, R.; Thomas, E.O.; Ramirez, P.; Miller, H.E.; Beckmann, A.; Zuniga, G.; Dobrowolski, R.; Frost, B. Pathogenic Tau Causes a Toxic Depletion of Nuclear Calcium ll ll Pathogenic Tau Causes a Toxic Depletion of Nuclear Calcium. Cell. Rep. 2020, 32, 107900. [Google Scholar] [CrossRef]
- Huang, Y.; Weisgraber, K.H.; Mucke, L.; Mahley, R.W. Apolipoprotein E: Diversity of cellular origin, structural and biophysical properties, and effects in Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23, 189–204. [Google Scholar] [CrossRef]
- Grehan, S.; Tse, E.; Taylor, J.M. Two Distal Downstream Enhancers Direct Expression of the Human Apolipoprotein E Gene to Astrocytes in the Brain. J. Neurosci. 2001, 21, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Ignatius, M.J.; Gebicke-haerter, P.J.; Pitasb, R.E.; Shootera, E.M. Apolipoprotein E in nerve injury and repair. Prog. Brain. Res. 1987, 71, 177–178. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theendakara, V.; Peters-libeu, C.A.; Spilman, X.P.; Poksay, K.S.; Bredesen, D.E.; Rao, R.V. Direct Transcriptional Effects of Apolipoprotein. Eur. J. Neurosci. 2016, 36, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Südhof, T.C. APOE2, APOE3, and APOE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theendakara, V.; Patent, A.; Peters, C.A.; Philpot, B.; Flores, S.; Descamps, O.; Poksay, K.S.; Zhang, Q.; Cailing, G.; Hart, M.; et al. Neuroprotective Sirtuin ratio reversed by APOE4. Proc. Natl. Acad. Sci. USA 2013, 110, 18303–18308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, M.H.; Wen, H.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol Suppresses β-Amyloid Production by Activating A Disintegrin and Metalloproteinase 10 Via the Upregulation of SIRT1-Coupled Retinoic Acid Receptor-β. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020, 11, 5540. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Xu, H.; Bu, G. Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol. Brain 2013, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Bagyinszky, E.; Giau, V.; An, S.A. Transcriptomics in Alzheimer ’s Disease: Aspects and Challenges. Int. J. Mol. Sci. 2020, 21, 3517. [Google Scholar] [CrossRef]
- Verheijen, J.; Sleegers, K. Understanding Alzheimer Disease at the Interface between Genetics and Transcriptomics. Trends Genet. 2018, 34, 434–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Lu, A.; Craessaerts, K.; Pavie, B.; Frigerio, C.S.; Corthout, N.; Qian, K.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease ll Resource Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Herrmann, A.G.; Mcqueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldsta, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell. Rep. 2019, 29, 3592–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Wang, M.; Zhan, Y.; Zhang, J.; Yang, X.; Fu, S.; Bi, D.; Gao, F.; Shen, Y.; Chen, Z. Single-nucleus RNA sequencing reveals transcriptional changes of hippocampal neurons in APP23 mouse model of Alzheimer’s disease. Biosci. Biotechnol. Biochem. 2020, 84, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Magistri, M.; Velmeshev, D.; Makhmutova, M.; Faghihi, M.A. Transcriptomics Profiling of Alzheimer’s Disease Reveal Neurovascular Defects, Altered Amyloid-β Homeostasis, and Deregulated Expression of Long Noncoding RNAs. J. Alzheimer’s Dis. 2015, 48, 647–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Wan, Y.; Al-ouran, R.; Mangleburg, C.G.; Perumal, T.M.; Lee, T.V.; Allison, K.; Swarup, V.; Funk, C.C.; Gaiteri, C.; Allen, M.; et al. Resource Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell. Rep. 2020, 32, 107908. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]

{kind=link}
| Molecules | DNA Binding | Nuclear Function | Target Genes | References |
|---|---|---|---|---|
| AICD | yes | Transcription regulator | STMN1 KAI1 NEP GSK3β Pink-1 | [67] [66] [68,69,70,75] [72,74] [89] |
| Aβ | yes | Transcription regulator | APP BACE APOE IRP1 KAI1 | [90,93] [90] [90] [29] [29] |
| tau | yes | DNA protection Chromatin remodeling Genome stability | Unknown genes | [111,112] [107,115] [117] |
| APOEε4 | yes | Transcription regulator | APP SirT1 MADD ADNP COMMD6 1700 other genes | [135,136] [133,138] [133] [133] [133] [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Andrea, L.; Stringhi, R.; Di Luca, M.; Marcello, E. Looking at Alzheimer’s Disease Pathogenesis from the Nuclear Side. Biomolecules 2021, 11, 1261. https://doi.org/10.3390/biom11091261
D’Andrea L, Stringhi R, Di Luca M, Marcello E. Looking at Alzheimer’s Disease Pathogenesis from the Nuclear Side. Biomolecules. 2021; 11(9):1261. https://doi.org/10.3390/biom11091261
Chicago/Turabian StyleD’Andrea, Laura, Ramona Stringhi, Monica Di Luca, and Elena Marcello. 2021. "Looking at Alzheimer’s Disease Pathogenesis from the Nuclear Side" Biomolecules 11, no. 9: 1261. https://doi.org/10.3390/biom11091261
APA StyleD’Andrea, L., Stringhi, R., Di Luca, M., & Marcello, E. (2021). Looking at Alzheimer’s Disease Pathogenesis from the Nuclear Side. Biomolecules, 11(9), 1261. https://doi.org/10.3390/biom11091261